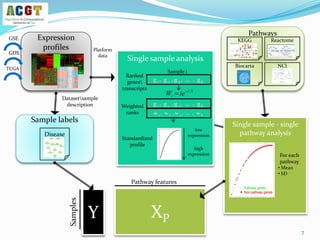
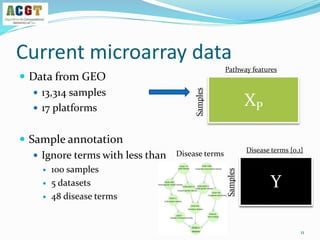
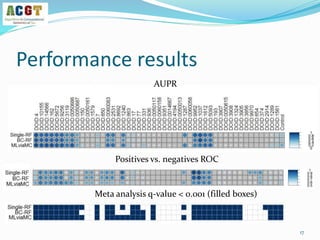
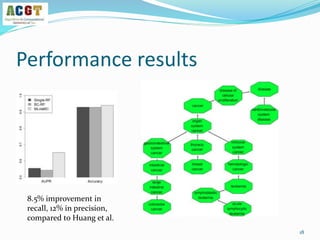
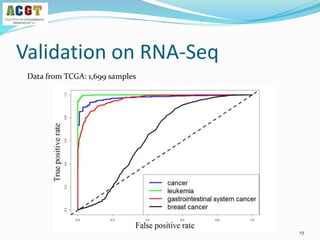
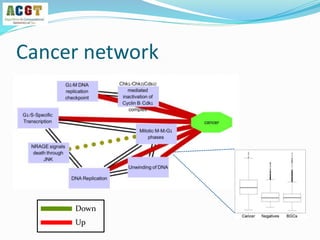
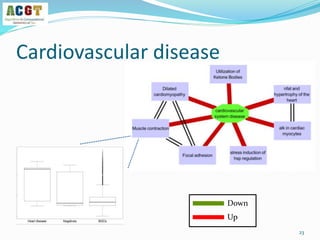
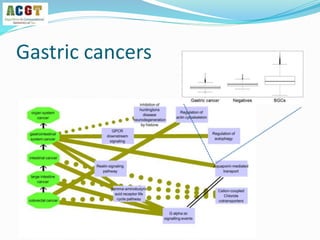
This document summarizes a study that integrated pathway and gene expression data from over 13,000 samples across 17 platforms to perform multi-label classification of 48 diseases. Pathway activity scores were calculated for each sample and used as features for classification, along with sample labels determined through manual dataset analysis. Classification was performed using multiple algorithms and validated through cross-validation and comparison to previous studies. Performance was improved over previous work, as shown by increased recall and precision. Relationships between diseases and pathways were also modeled in a network graph.












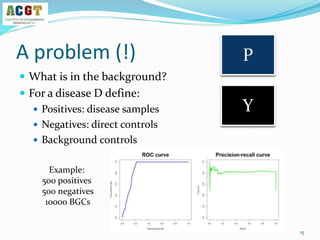
![How to validate an classifier?
Use leave-dataset out cross-validation
Global AUC scores: each prediction Pij vs the correct label Yij
Disease based AUC scores: consider each column separately
14
Y
Disease terms {0,1}
Samples
P
Probabilities [0,1]
Samples
The output of a multi-label learner
Test set](https://image.slidesharecdn.com/davidamarnetworksig2014-140721180426-phpapp02/85/NetBioSIG2014-Talk-by-David-Amar-14-320.jpg)














































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)




![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)







