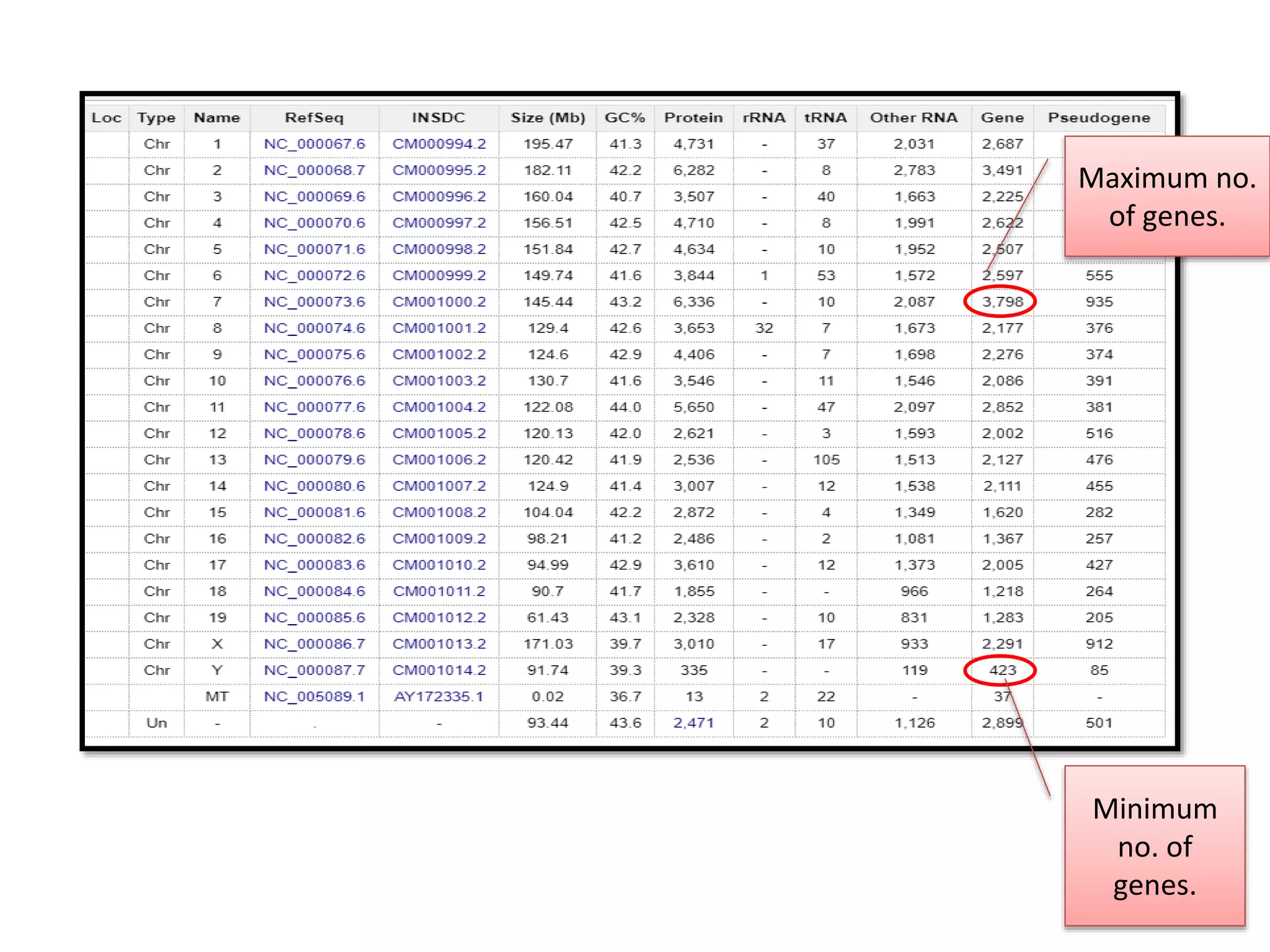
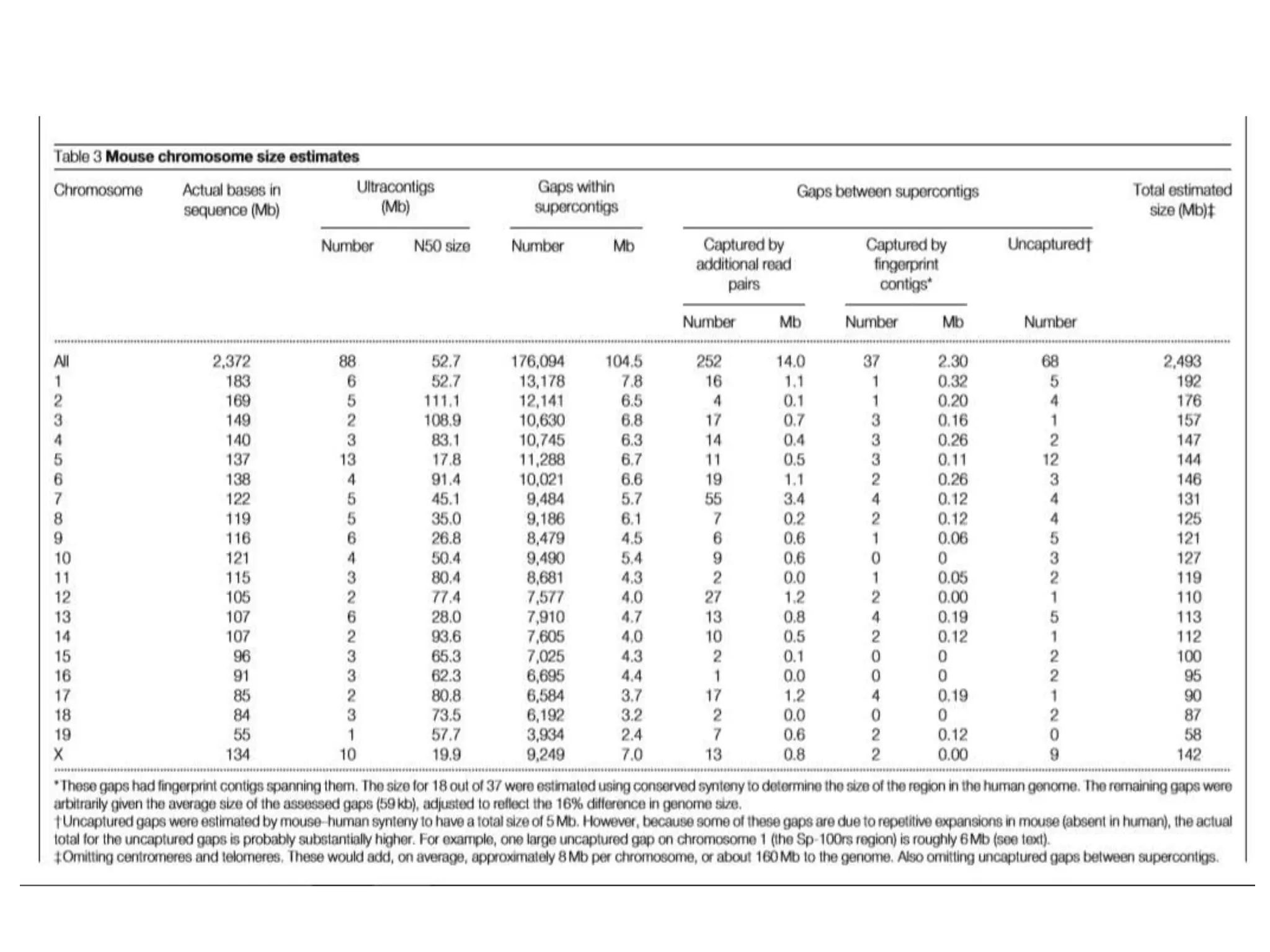
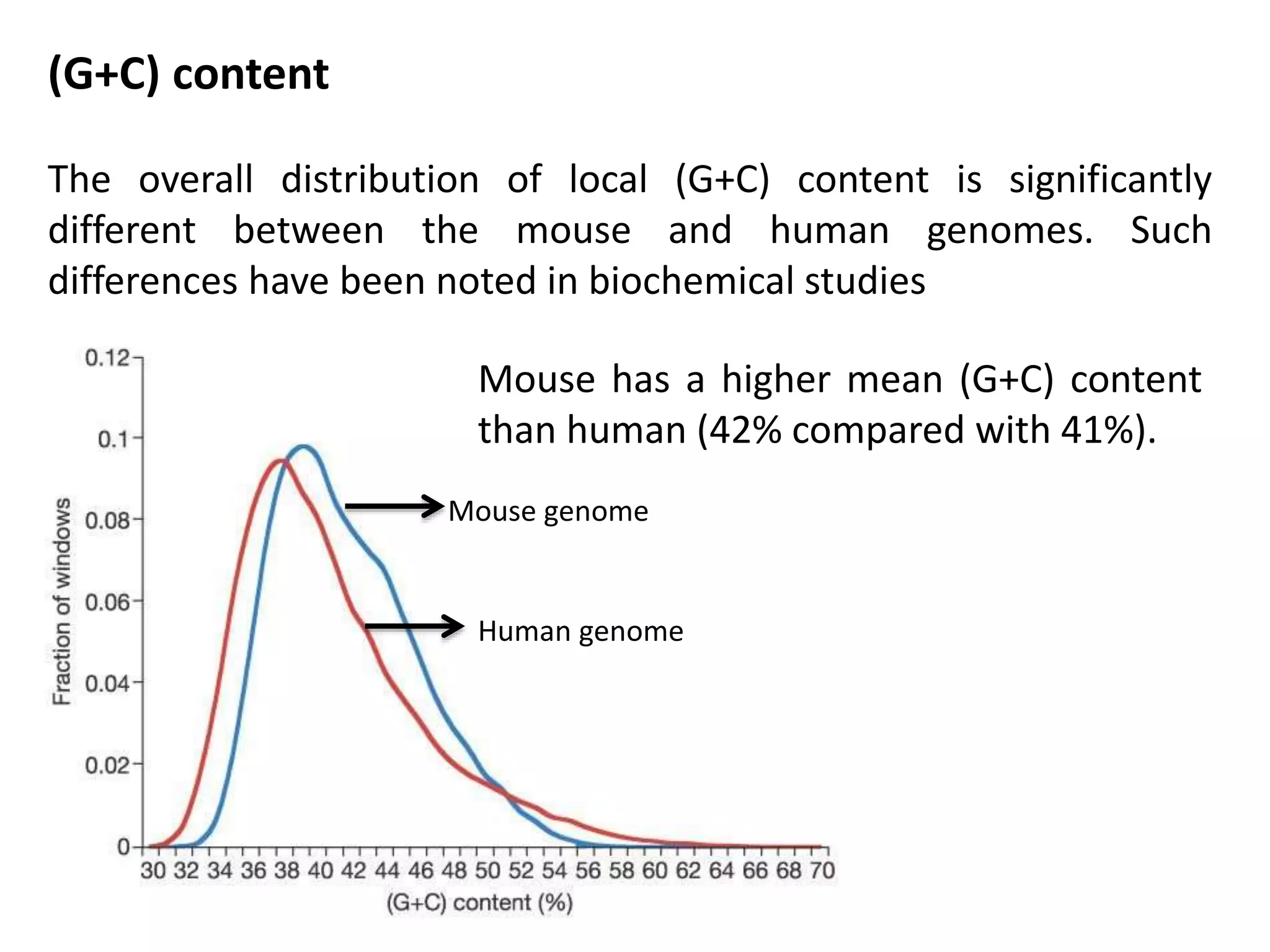
The document discusses the similarities and differences between the mouse and human genomes, including the role of mice as model organisms in genetic research due to their anatomical and genetic similarities to humans. It covers the mouse genome sequencing efforts, strategies used, and the findings related to conserved genetic regions and variations like SNPs between strains. The document highlights the implications of this research for understanding genetics, development, and evolution.