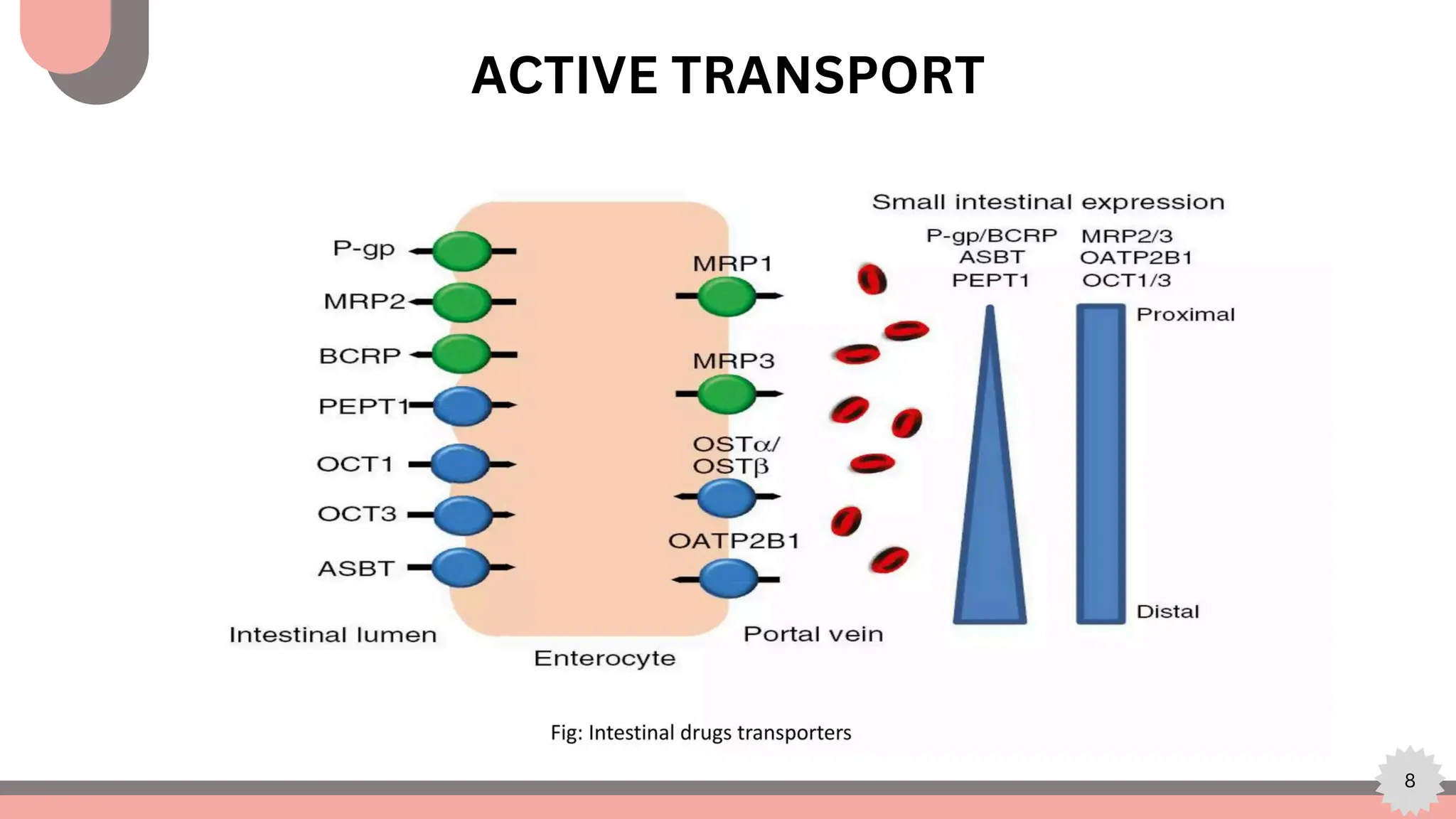
The document explores computational modeling techniques in drug development, focusing on drug excretion and the role of active transporters like P-glycoprotein (p-gp), breast cancer resistance protein (BCRP), and various nucleoside transporters. It highlights the importance of these transporters in pharmacokinetics, specifically how they affect drug absorption, distribution, and elimination. Additionally, the document details modeling efforts to create pharmacophore and QSAR models aimed at improving predictions related to drug disposition and enhancing drug bioavailability.








![11
Ekins and colleagues generated 5 computational pharmacophore models to predict the
inhibition of P-gp from in vitro data on a diverse set of inhibitors with several cell systems.
It includes inhibition of digoxin transport and verapamil binding in Caco-2 cells; vinblastine
and calcein accumulation in P-gp expressing LLC-PK1(L-MDR1) cells [cell exhibiting
epithelial morphology was isolated from kidney of male 3-4 week pig] and vinblastine
binding in vesicles derived from CEM/VLB 100 cells[cellosaurus cell line- is a cytogenetic
characterization of chromosomal rearrangement in human vinblastine resistant CEM cell
line]
By comparing and merging all P-gp pharmacophore models, common areas of identical
chemical features as well as their geometric arrangement were identified to the substrate
requirements for P-gp.](https://image.slidesharecdn.com/modellingtechniques-240811041251-5962fe03/75/MODELLING-TECHNIQUES-in-Computer-Aided-drug-development-11-2048.jpg)


























































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)








