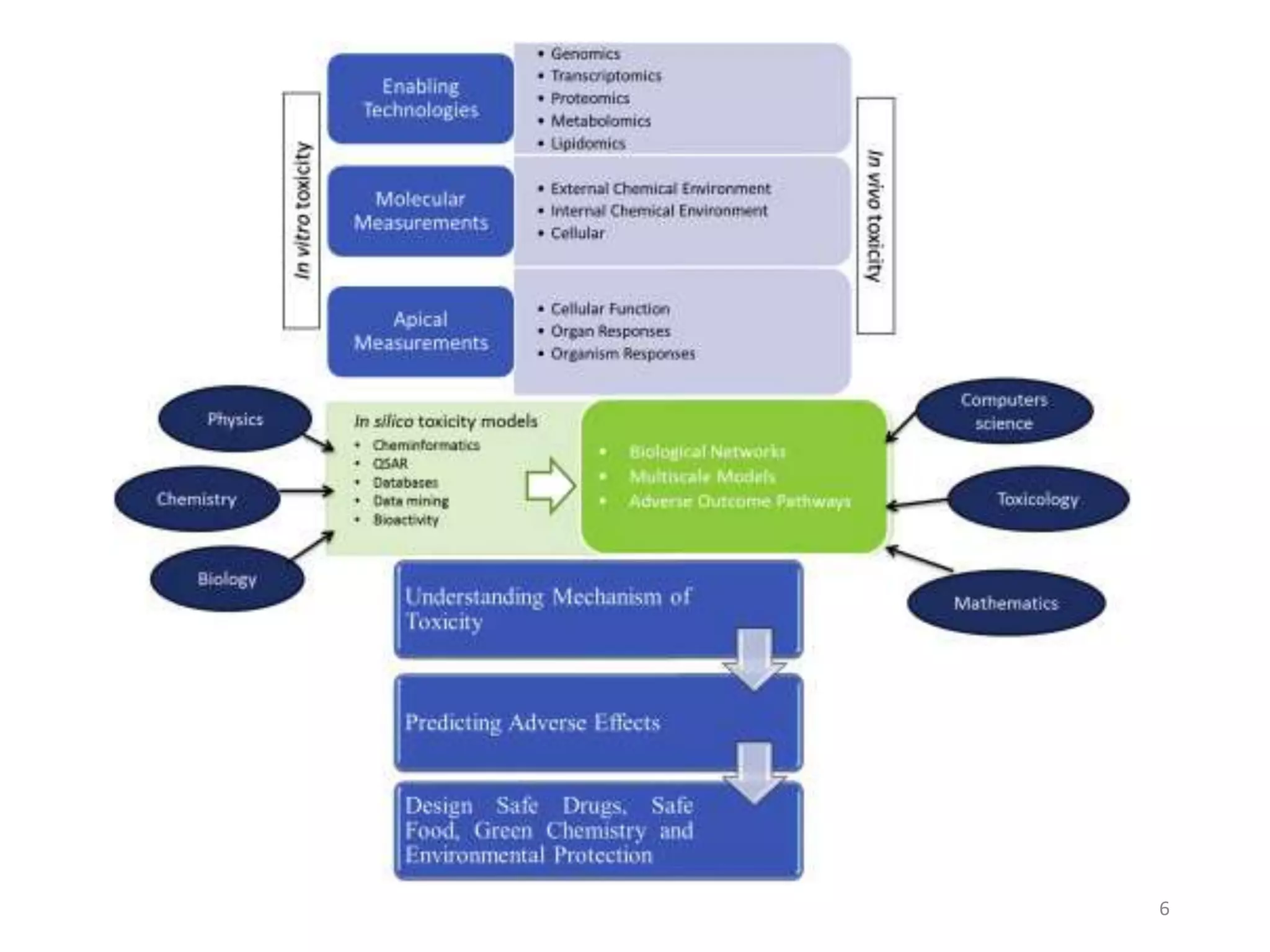
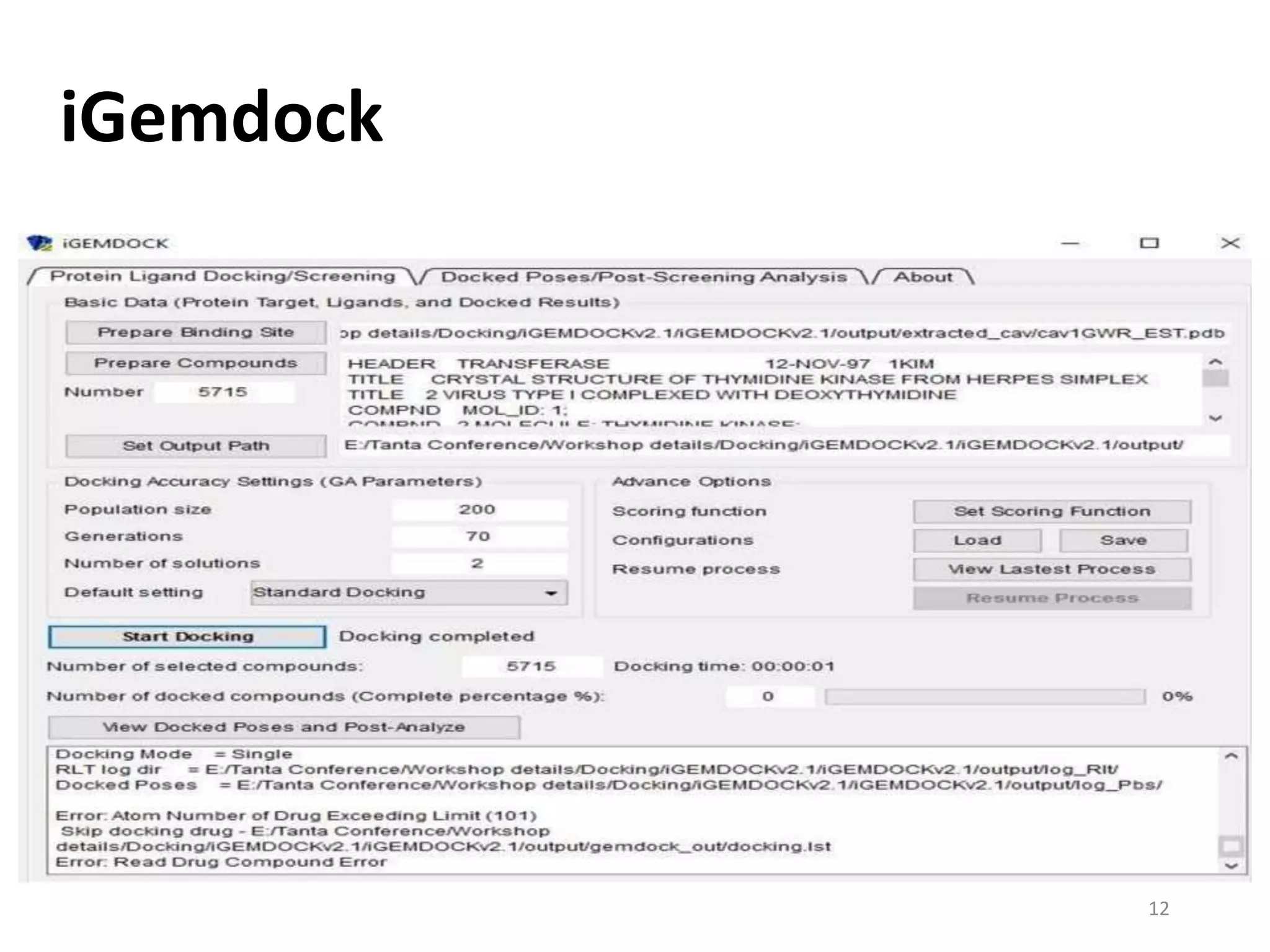
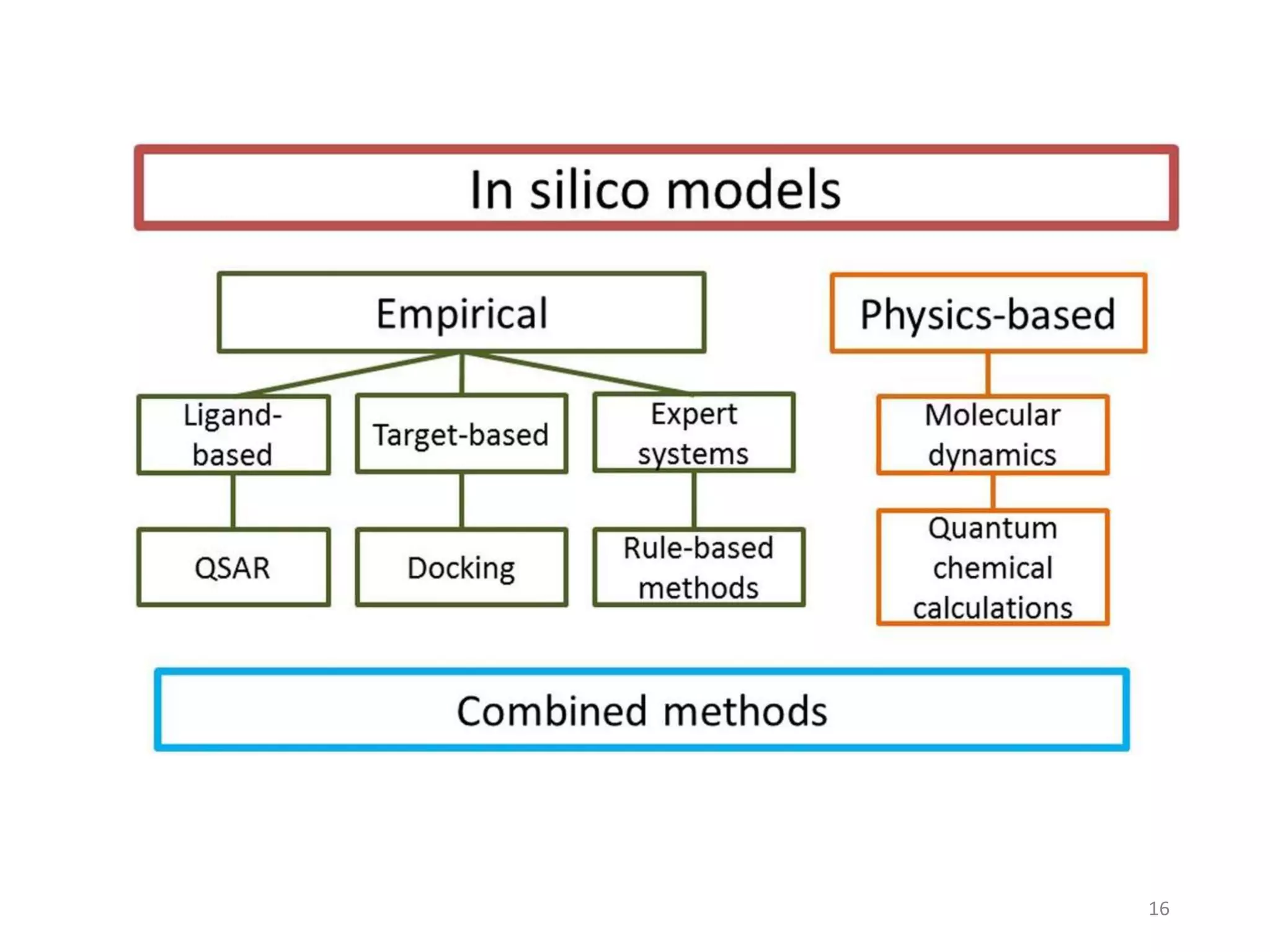
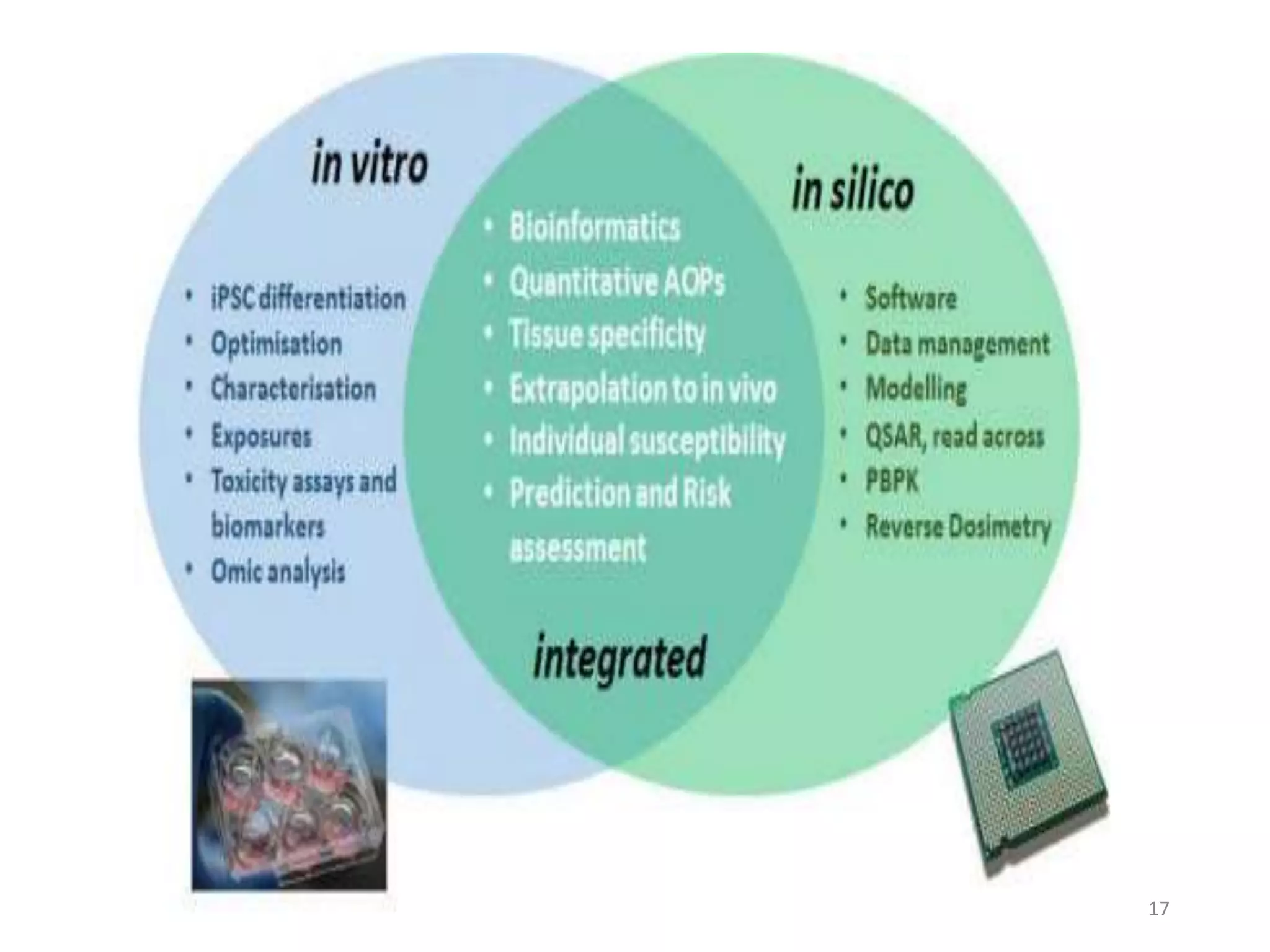
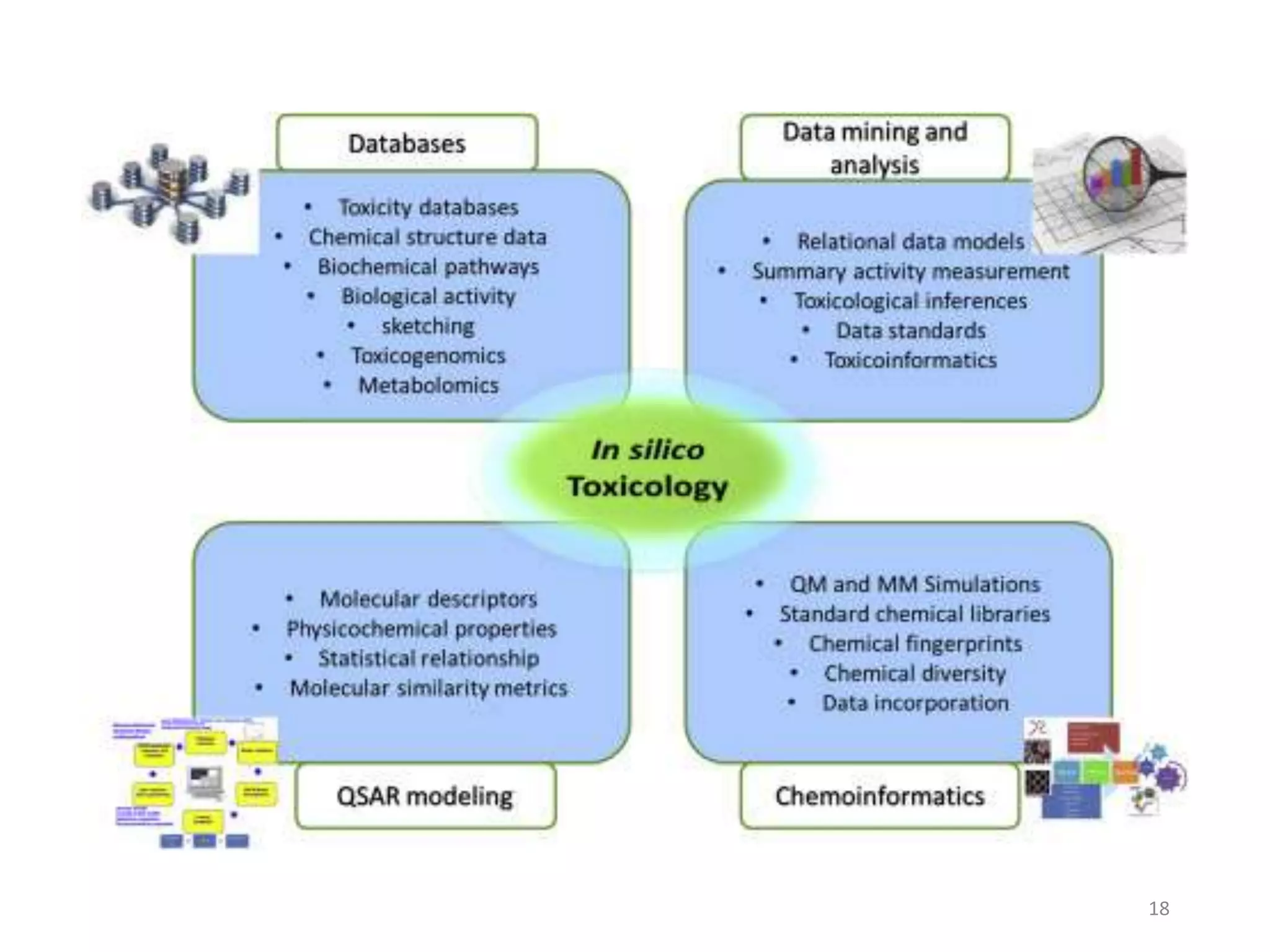
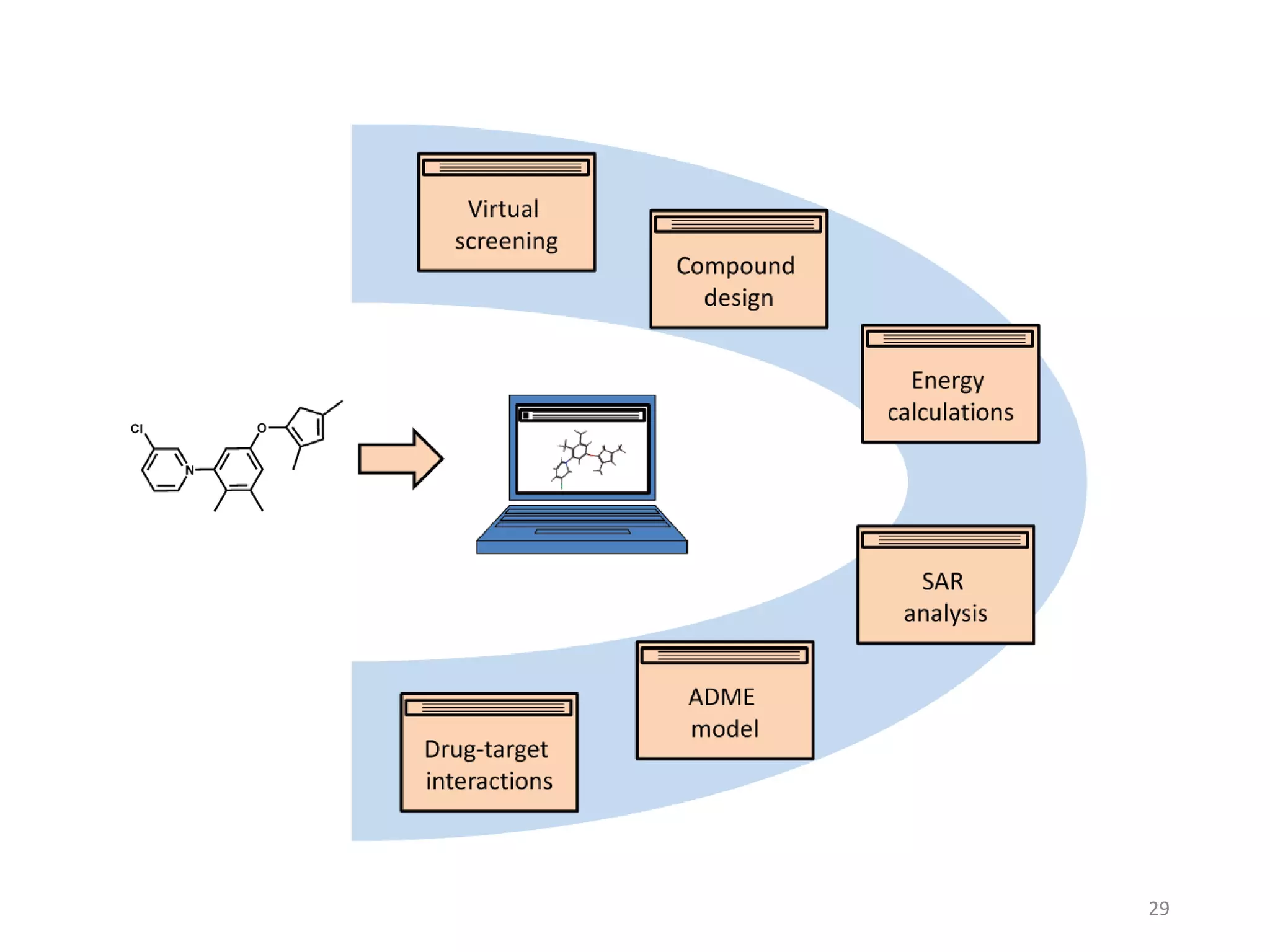
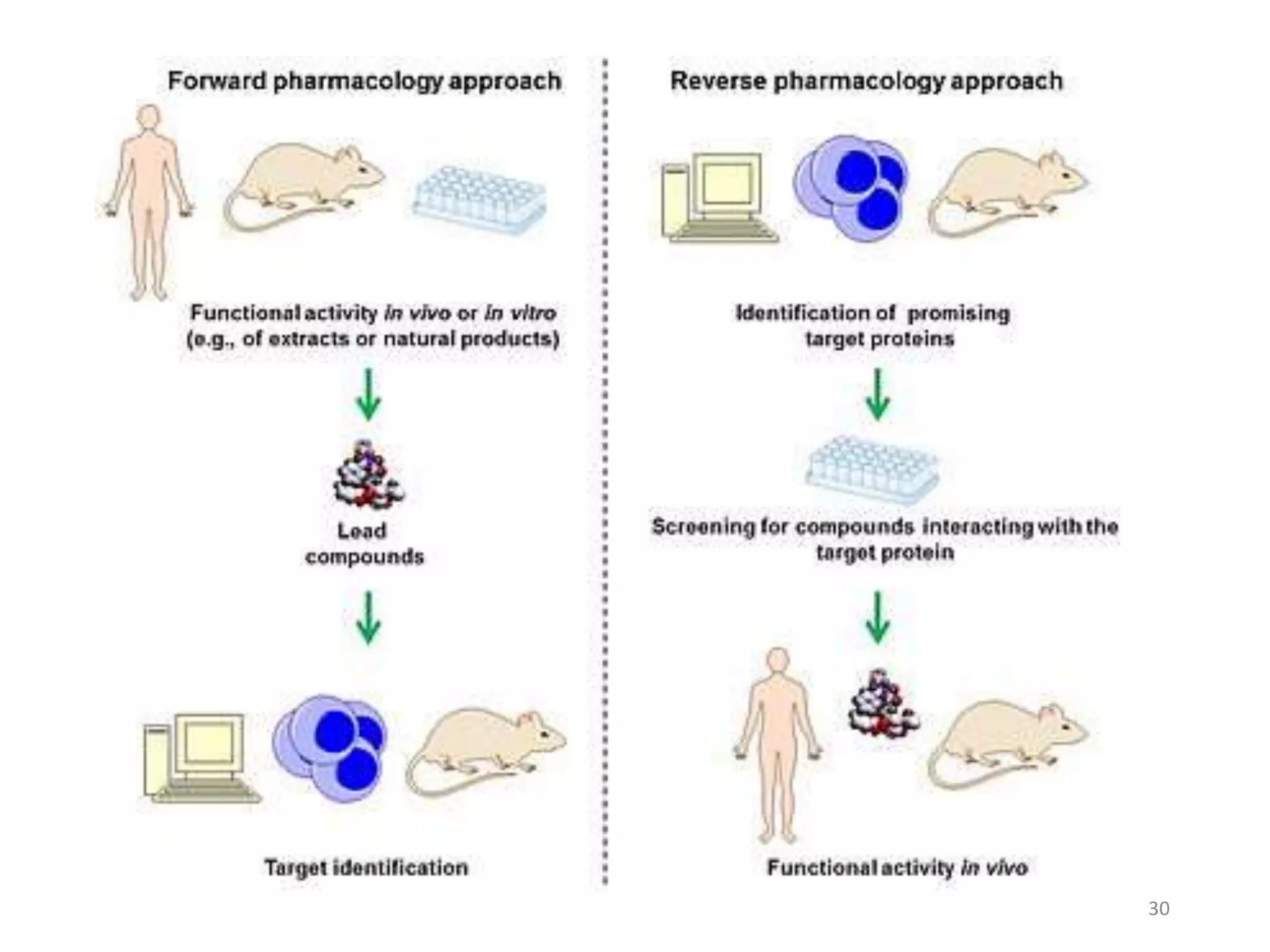
This document summarizes computational modeling techniques for predicting drug absorption, distribution, and excretion properties. It discusses both quantitative and qualitative modeling approaches. For absorption, it describes models for predicting solubility, permeability, and factors like ionization state and transporters. For distribution, it covers volume of distribution, plasma protein binding, and blood brain barrier permeability modeling. For excretion, it discusses challenges modeling clearance but focuses on estimating clearance from in vitro data. The document provides examples of specific modeling tools and considers limitations of current approaches.
























































![Computational modeling in_drug_disposition[1]](https://cdn.slidesharecdn.com/ss_thumbnails/computationalmodelingindrugdisposition1-200604161524-thumbnail.jpg?width=640&height=640&fit=bounds)


































