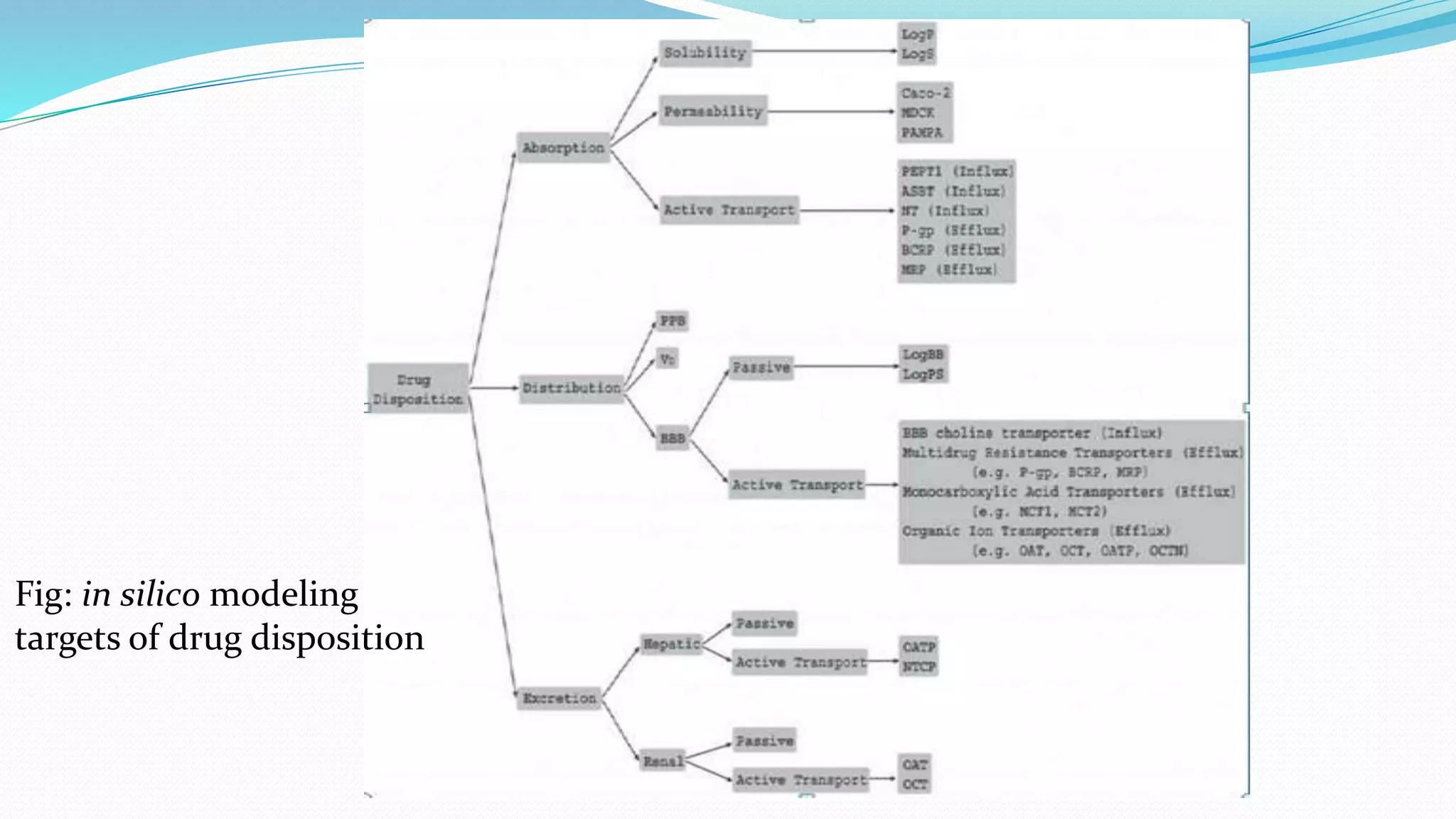
This document discusses in silico modeling techniques for predicting absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of drug candidates. It describes quantitative approaches like pharmacophore modeling and docking studies, as well as qualitative approaches like quantitative structure-activity relationship (QSAR) and quantitative structure-property relationship (QSPR) studies. Specific techniques are discussed for modeling various ADMET properties like solubility, permeability, plasma protein binding, blood-brain barrier penetration, and clearance. Transporters, ionization, and data quality are also mentioned as important factors. Commercial software packages are noted that can simulate these processes.






























































![Computational modeling in_drug_disposition[1]](https://cdn.slidesharecdn.com/ss_thumbnails/computationalmodelingindrugdisposition1-200604161524-thumbnail.jpg?width=640&height=640&fit=bounds)


























