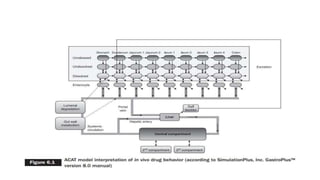
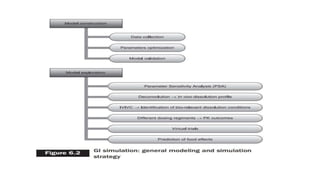
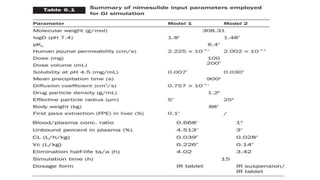
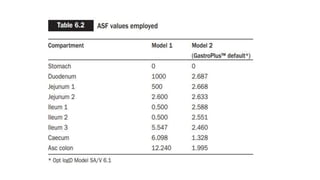
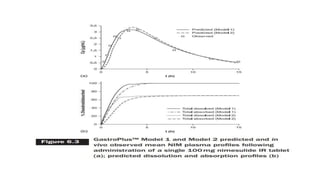
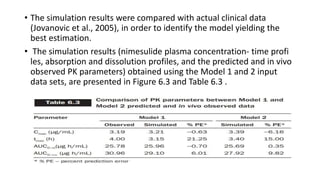
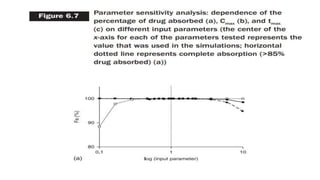
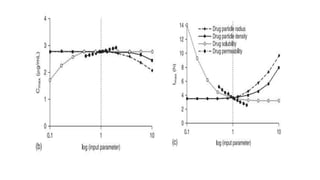
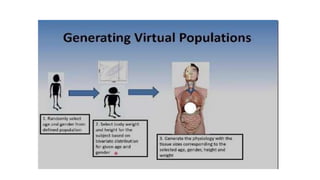
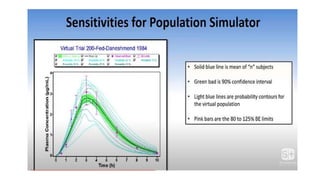
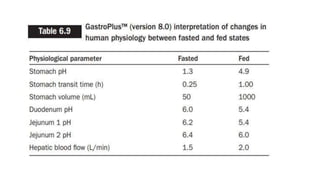
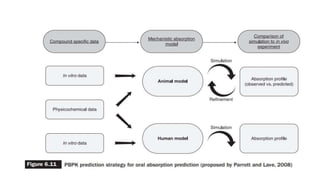
The document discusses the role of computer-aided biopharmaceutical characterization in simulating gastrointestinal absorption for drug development, emphasizing the importance of in silico models in predicting drug absorption under various conditions. It reviews different models, such as the advanced compartmental absorption and transit (ACAT) model and software tools like GastroPlus, detailing how they integrate physiological data to improve predictions of drug pharmacokinetics. Furthermore, the document highlights the process of model construction using different input parameters, showcasing a case study on nimesulide to illustrate the impact of assumptions on model accuracy.






































































![LEGAL PROTECTION OF IN[1] CADD pharmaceutics](https://cdn.slidesharecdn.com/ss_thumbnails/legalprotectionofin1-240320035446-be6dd039-thumbnail.jpg?width=640&height=640&fit=bounds)



























































