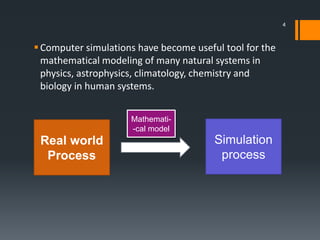
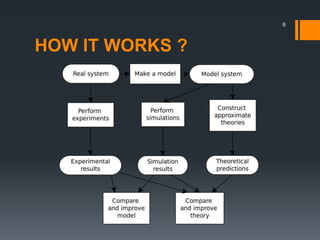
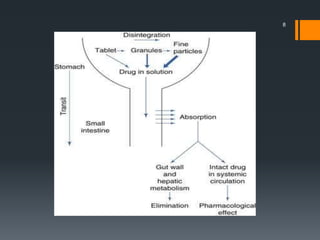
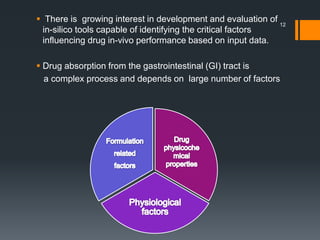
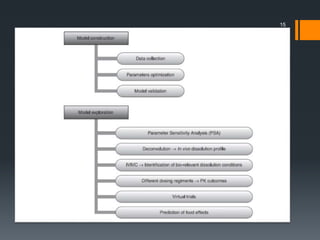
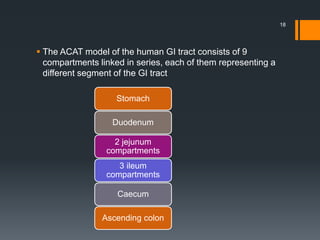
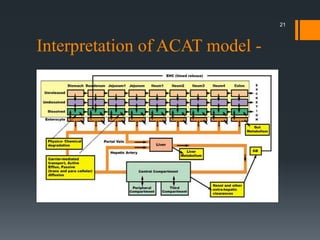
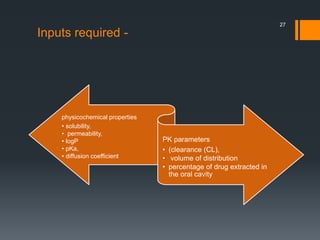
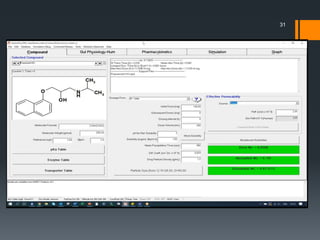
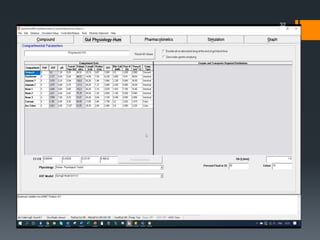
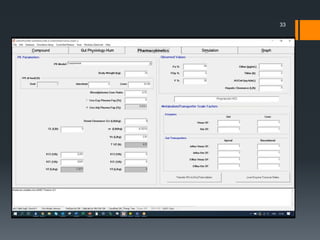
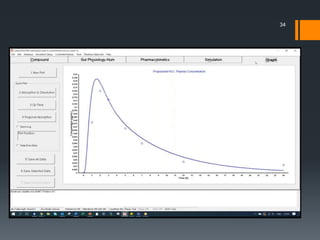
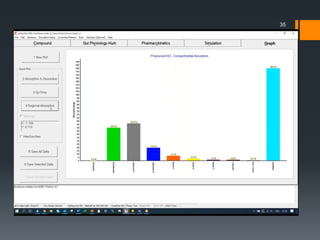
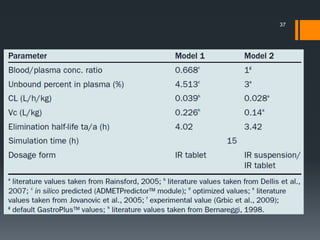
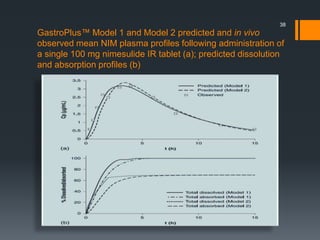
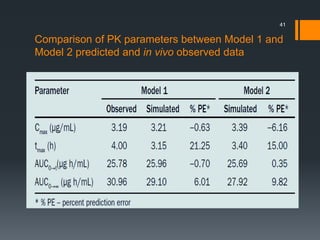
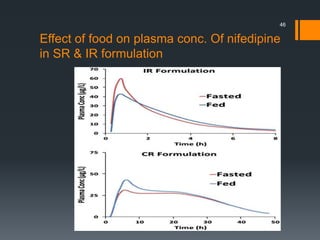
The document discusses a gastric absorption simulation developed by Sagar S. Bhor, aimed at understanding the drug absorption process from the gastrointestinal (GI) tract. It highlights the importance of in-silico simulations in predicting drug behavior, including the use of models like the advanced compartmental absorption and transit (ACAT). Additionally, it details the GastroPlus software's functionality in modeling drug absorption and its advantages over traditional in vivo studies.