Computers have been used in pharmaceutical research and development since the 1940s. Early computers were large mainframe systems that were expensive and shared between organizations. By the 1960s, some pharmaceutical companies had acquired early computers like the IBM 650 to assist with scientific tasks. Today, computers are essential for tasks across the pharmaceutical industry from drug design and clinical trials to manufacturing, sales, and more. Advanced statistical modeling and software continue to be important tools in pharmaceutical research and development.





























![NON-PARAMETRIC METHODS
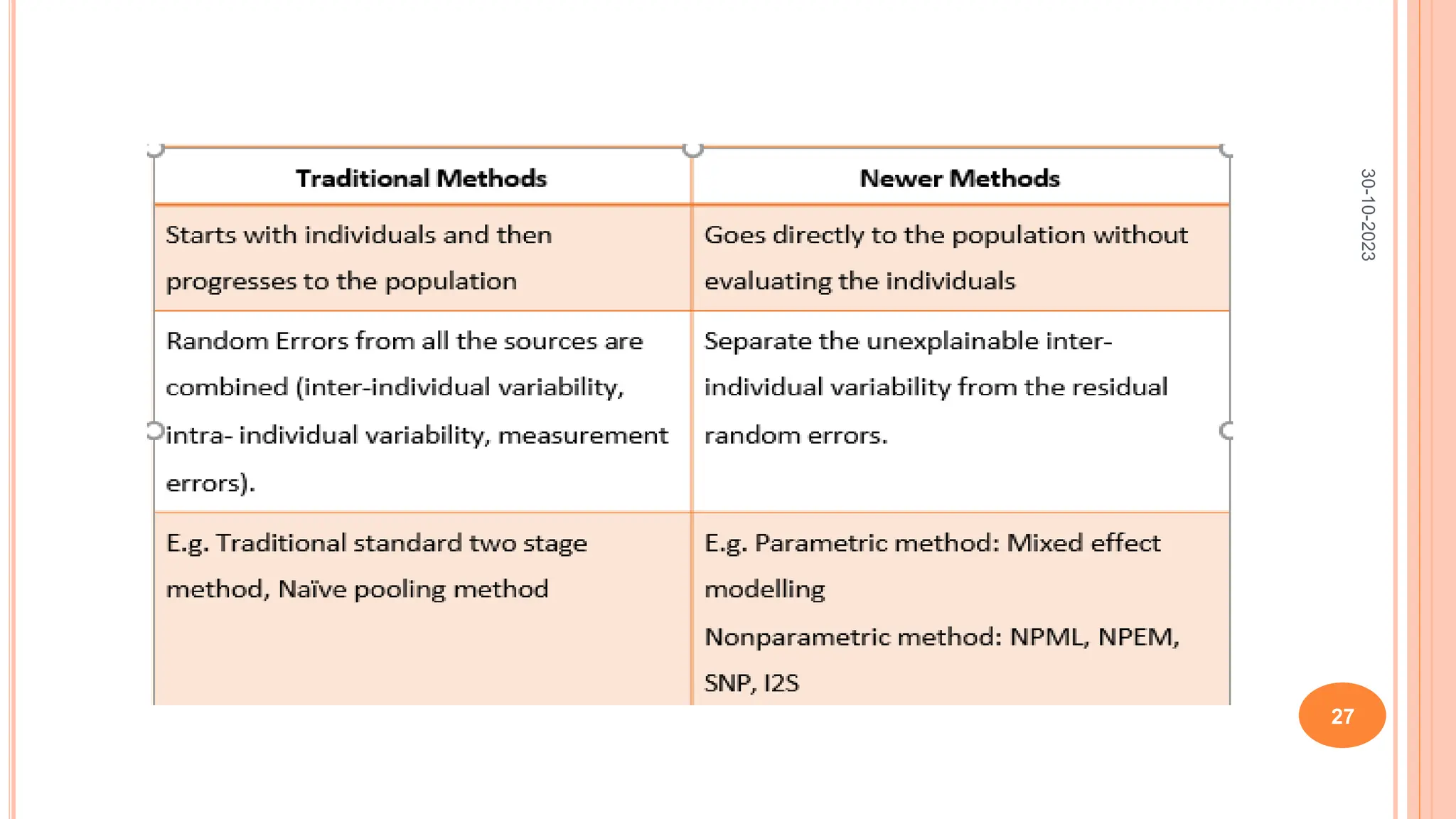
Non parametric methods do not assume any specific distribution of parameters
about the population values, but rather allow for many possible distributions.
In this method, the entire population distribution of each parameter is
estimated from the population data.
This permits visual inspection of distribution before committing to one.
Different non parametric methods are:
Non parametric maximum likelihood [NPML]
Non parametric expectation maximization [NPEM]
Nonparametric Semi/ smooth non parametric method [SNP]
30-10-2023
31](https://image.slidesharecdn.com/hisoryofcomputersinpharmaceuticalresearchpresentation-231030050509-ae2e62f2/75/hisory-of-computers-in-pharmaceutical-research-presentation-pptx-31-2048.jpg)
![NON PARAMETRIC MAXIMUM LIKELIHOOD [NPML]
This method permits all forms of distributions including those containing sharp
changes, such as discontinuities and kinks.
It uses maximum likelihood as estimator.
NON PARAMETRIC EXPECTATION MAXIMIZATION [NPEM]
This method is preferred to any parametric method when there is an unexpected
multimodal or non-normal distribution of at least one of the nodal parameters.
It eliminates the need for initial guesses which are required for nonlinear least
square procedure. It is preferable to traditional method in case of sparse data. It uses
expectation maximization as the estimator.
SEMI/ SMOOTH NON PARAMETRIC METHOD [SNP]
This method places some restrictions on the type of parametric distributions considered.
Functions that are not permitted include those containing sharp edges and discontinuities.
30-10-2023
32](https://image.slidesharecdn.com/hisoryofcomputersinpharmaceuticalresearchpresentation-231030050509-ae2e62f2/75/hisory-of-computers-in-pharmaceutical-research-presentation-pptx-32-2048.jpg)

























































































![COMPUTATIONAL FLUID DYNAMICS [Autosaved] (1).pptx](https://cdn.slidesharecdn.com/ss_thumbnails/computationalfluiddynamicsautosaved1-241225092723-6910a5cb-thumbnail.jpg?width=640&height=640&fit=bounds)


































![Rheumatic Fever CASE PRESENTATION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationautosaved-251123182512-9d9b0da4-thumbnail.jpg?width=640&height=640&fit=bounds)

