Downloaded 22 times












![ Common neutralizingagents and methodsincludetheaddition ofpolysorbate, the addition oflecithin,and/or
dilutionmethods(1).
USP <61>states thatifno suitableneutralizingmethodcanbefound, it can beassumedthat thefailureto
isolate the inoculatedmicroorganismsis attributable to themicrobialactivityoftheproduct(1).Proceed by
performing the test with thehighest dilution factorcompatiblewithmicrobial growthandthespecific
acceptancecriterion in caseothermicroorganisms are notinhibitedby the product (1). USP <62>continues on
to say thatfora given product,ifthe antimicrobial activitywithrespect to a microorganismforwhich testing is
prescribedcannot beneutralized, then it is to beassumedthat theinhibited microorganismwillnotbepresent
in the product(10).Most companiescontinuein themethod developmenttrialsuntil a suitablemethod is
identified.
Dependingon thenature ofthe product andtherequiredlimitofmicroorganismsallowed,choose the
appropriatemethodto usein the method development trials. Again, themethodsincludethe membrane
filtration method,theplatecountmethod, andtheMPN method.
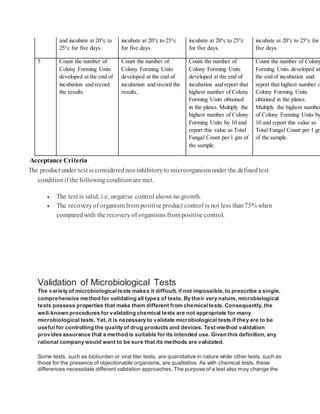
For the membranefiltrationmethod, filtration mustbeperformed with filtersthat havea poresizenot greater
than 0.45m(1).Thetypeoffiltermaterial is chosenin such a way that the bacteria-retainingefficiencyis not
affectedby the componentsofthesample (1). Common filtermaterialsinclude cellulose, nylon, and
Polyvinylidene fluoride(PVDF).
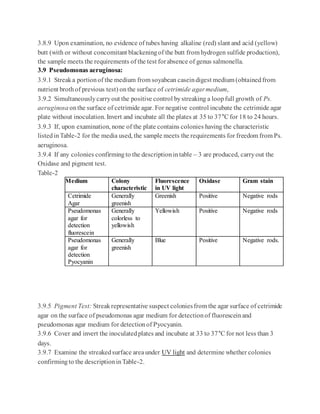
Membrane Filtration Example
1. Transfera suitablequantityofthesampleprepared(preferablyrepresenting 1 gram[g] oftheproduct) to the
membraneandfilterimmediately. Rinsethefilterwithan appropriatediluent(1).
2. For Total Aerobic Microbial Counts (TAMC),transferthefilter to soybean-caseindigest agar.
3. For Total YeastMicrobial Counts (TYMC),transfer the filterto sabourauddextroseagar.
4. Incubate alloftheplatesaccordingto the compendialguidance(1).
There aretwo methods listedin the compendialchaptersfortheplatecountmethods.They arethepourplate
methodandthesurface-spreadmethod.
Pour Plate MethodExample:
1. Add 1 milliliter (mL) ofthesamplepreparationto duplicatepetri dishes.
2. Cover the sample with 15-20 mL ofmolten media (cooled to ~45C).
3. Allowtheplatesto solidify atroomtemperature.
4. Inverttheplates andincubateaccordingto thecompendial guidance(1).
Spread Plate MethodExample:
1. Add media to sterilepetridishes andallowthemedia to solidify.
2. Add a measured amountofnotless than0.1 mL oftheprepared sampleto duplicatesolidifiedmedia plates.
Spreadthesampleoverthemedia surface.
3. Inverttheplates andincubateaccordingto thecompendial guidance(1).](https://image.slidesharecdn.com/mlt-161208025230/85/Mlt-18-320.jpg)




![For many companies, the question ofhow to treatclinical productsverses commercial productsarisesin
meeting rooms.Forcommercial products, most companies agreethat theindustry practiceis to validatethree
lots at a minimumfor method validations.
What about clinical products?Clinical productsarea differentstory. Itis difficultsometimes to havethree lots
ofproductin orderto performa completevalidation.Smallamountsofproduct areoftenmadeforusein
clinics.Formulations change frequently depending on fieldstudies anddevelopmentsdiscoveredthroughout
the early phases ofa project. Itis acceptableto perform method validation ononelot forclinical products.
Chances are thecompany will beredeveloping andrevalidatingwitheach formulation changeoftheproduct. It
may take yearsfora product to reach commercial launch.
Keep an eyeontheproduct, however.The moment theproduct moves to commercial manufacturing,a three-
lot validation needs to occur.
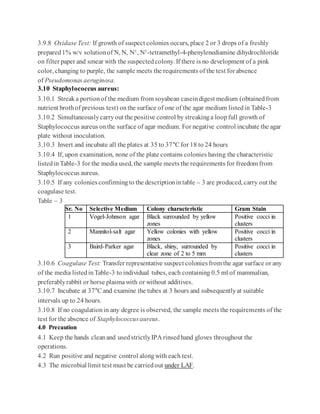
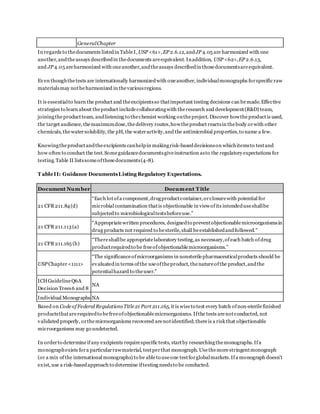
Validationacceptancecriteriais essentialwhen determining ifa methodwas properlyvalidated.When verifying
the suitability ofthemembranefiltration or the Plate-Count Method,“a mean count ofany ofthetest
organismsnotdifferingby a factorgreater than2 fromthevalue ofthe control in theabsenceofproductmust
be obtained”(1).In other words,thepercent recoverybetweentheinoculatedproductdilutions andthepositive
controls mustbeat least50%.Somecompanies usean internal70%recovery criterionthat is acceptable
becauseit is morestringent.
The compendial chaptersgiveadditional detailto the validation criteriafortheMPN method.“When verifying
the suitability oftheMPN Method,thecalculatedvaluefromtheinoculums mustbewithin95%confidence
limits oftheresults obtainedwith thecontrol”(1). Ifthe criterioncannotbe metfor oneormore ofthe
organismstestedwith any ofthedescribedmethods,themethodand testconditions that comeclosestto the
criteria areusedto testtheproduct(1).
Specifiedmicroorganismsmustbe recovered duringthevalidationfortheassayto bevalid. Thetiterplates
must demonstratenotmorethan 100 cfuwas utilizedto achievethepositiveresults (10).
Aftervalidations arecomplete,routine testing maybegin. Followingvalidation activities, reportsareusually
written to approvethestudies andmethods (or standard operating procedures[SOPs]) arewritten to lock down
the way thetests are routinelyperformed.
Routinetesting fortheHMLTwill containtheTAMC, the TYMC, and any specified microorganismchallenges.
All representativecolonies ofgrowth obtainedfromany portionofthetest needsto be identified to thespecies
level ifpossible. Themicroorganismsneedto be researched anddeterminedifthey areobjectionable
microorganisms.
There is a regulatoryexpectation that recovered microorganismsareidentifiedfrom nonsterileproducts.There
havebeenwarningletters issuedto companies failingto comply withthisexpectation.Product recallshavealso
occurred.
In orderto determineifa microorganismis objectionable, USP <1111>provides information to aide in therisk
assessment.Themicroorganisms’ significanceshouldbe evaluated by researchingthefollowing:
Number ofmicroorganisms (1 cfuor1X106
cfu)](https://image.slidesharecdn.com/mlt-161208025230/85/Mlt-23-320.jpg)







This document provides procedures for conducting a Microbial Limit Test (MLT). The test involves several steps: sample pretreatment, total aerobic count using membrane filtration or plate count methods, and examination for specified microorganisms like E. coli, Salmonella, Pseudomonas aeruginosa, and Staphylococcus aureus. Positive and negative controls are run alongside each test. The procedures describe preparing bacterial and fungal suspensions, inoculating various media, and incubating and examining plates to identify microbial growth or absence. Safety precautions like using clean gloves and running tests under laminar airflow are also outlined.








































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)







![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)





