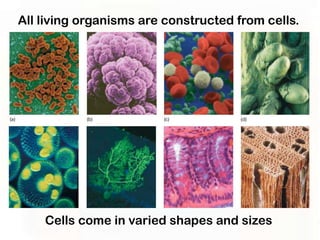
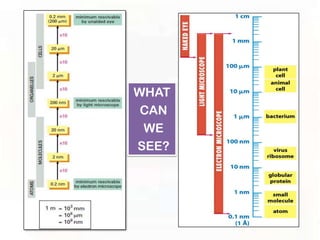
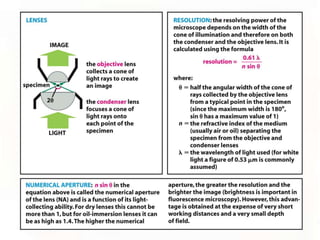
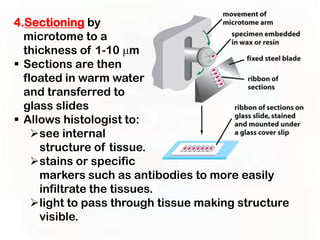
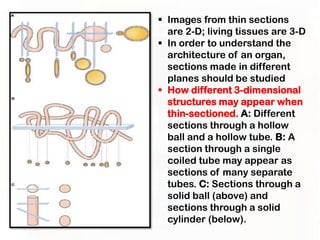
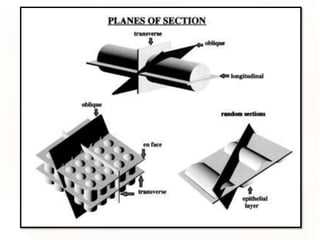
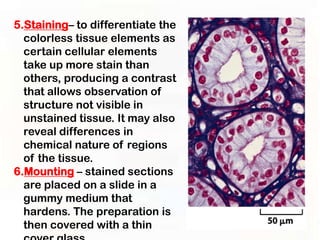
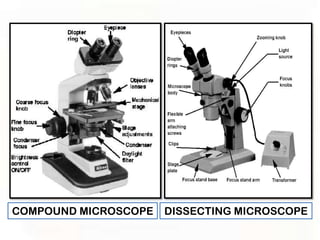
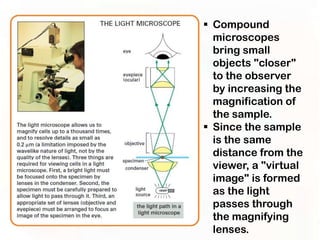
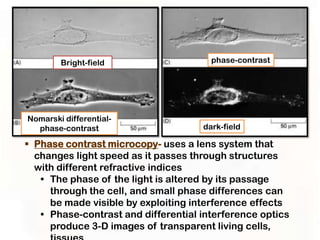
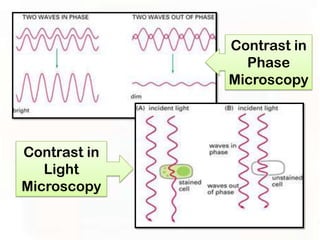
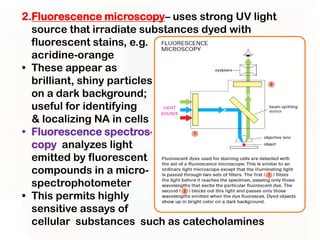
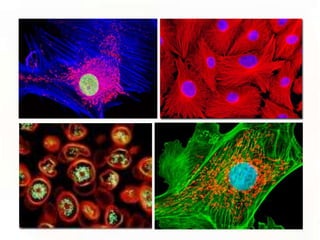
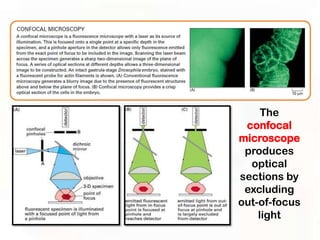
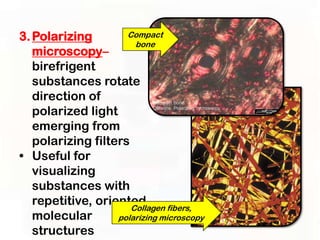
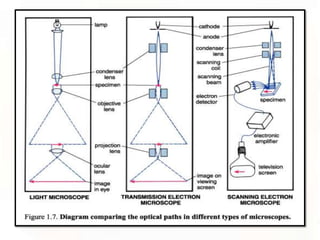
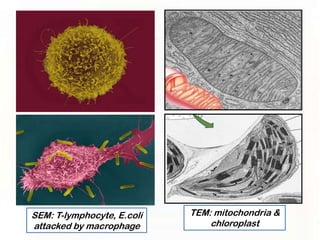
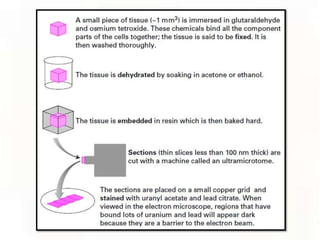
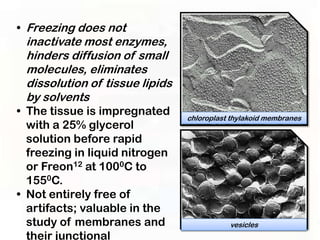
This document provides an overview of microscopic anatomy and various microscopy techniques. It discusses that [1] cells are the basic building blocks of living organisms and come in varied shapes and sizes, [2] microscopy involves using probes like light or electron beams that interact with tissue components to produce images, and [3] important considerations in microscopic analysis include the probe size and its ability to interact with and observe the object being investigated. It then describes various microscopy methods like light, fluorescence, polarization, and electron microscopy as well as tissue preparation techniques and important microscopy terms.