Downloaded 343 times






![To encompass most pharmaceutical powders ranging in size 1 - 120 mm:
- Blockage of electrical conductivity path (Coulter-counter)
- Light blockage (HIAC) [adopted by USP]
- Light scattering (Royco)
- Laser scattering (Malvern)
3.1.4 Other techniques
- Centrifugation
- Air suspension
- Sedimentation (Andersen pipette)
Common Techniques for Measuring Fine Particles of Various Sizes
3.2 Determination of Surface Area
Grinding operation:
particle size ==> surface area.
Brunauer-Emmett-Teller (BET) theory of adsorption
Most substances will adsorb a monomolecular layer of a gas under certain conditions of
partial pressure (of the gas) and temperature.
Knowing the monolayer capacity of an adsorbent (i.e., the quantity of adsorbate that can be
accommodated as a monolayer on the surface of a solid) and the area of the adsorbate molecule, the
surface area canbe calculated.
4. SOLUBILITY
Solubility > 1 % w/v
=> no dissolution-related absorption problem
Highly insoluble drug administered in small doses may exhibit good absorption
Unstable drug in highly acidic environment of stomach, high solubility and consequent rapid
dissolution could result in a decreased bioavailability](https://image.slidesharecdn.com/fullip1-151104153439-lva1-app6892/85/Industrial-Pharmacy-Notes-for-M-Pharmacy-6-320.jpg)

![where
D = drug molecule
C = complexing agent (ligand)
St = total solubility of free drug [D] and the
drug in the complex [DxCy]
Ligand (Complexing Agents)
- Vitamin K - Caffeine
- Menadione - Benzoic acid
- Cholesterol - PEG series
- Cholate salt - PVP
- b-cyclodextrin](https://image.slidesharecdn.com/fullip1-151104153439-lva1-app6892/85/Industrial-Pharmacy-Notes-for-M-Pharmacy-9-320.jpg)

![ The rate and extent of absorption decreased with the increasing polarity of molecules.
Partition coefficient (distribution coefficient): the ratio in which a solute distributes itself
between the two phases of two immiscible liquids that are in contact with each other (mostly
n-octanol/water).
6.2 Ionization Constant
The unionized species are more lipid-soluble and hence more readily absorbed.
The GI absorption of weakly acidic or basic drugs is related to the fraction of unionized drug
in solution.
Factors affecting absorption:
- pH at the site of absorption
- Ionization constant
- Lipid solubility of unionized species
“pH-partition theory”
Henderson-Hasselbalch equation
For acids:
pH = pKa + log [ionized form]/[unionized form]
For bases:
pH = pKa + log [unionized form]/[ionized form]
Determination of Ionization Constant
1. Potentiometric pH-Titration
2. pH-Spectrophotometry Method
3. pH-Solubility Analysis](https://image.slidesharecdn.com/fullip1-151104153439-lva1-app6892/85/Industrial-Pharmacy-Notes-for-M-Pharmacy-11-320.jpg)






![ The intermolecular theory.
The liquid surface film theory.
THE MECHANICAL THEORY:
It occurs between irregularly shaped particles. Also increases the number of contact points between the
particles. The mechanical theory proposes that under pressure the individual particles undergo
elastic/plastic or/& brittle deformation & that the edges of the particles intermesh deforming a mechanical
bond. If only the mechanical bond exists, the total energy of compression is equal to the sum of the energy
of deformation, heat & energy absorbed for each constituent. Mechanical interlocking is not a major
mechanism of bonding in pharmaceutical tableting.
INTERMOLECULAR THEORY:
The molecules [or ions] at the surface of solids have unsatisfied forces [surface free energy] which interact
with the other particles in true contact. Under pressure the molecules in true contact between new clean
surfaces of the granules are close enough so that vanderwals forces interact to consolidate the particles.
Materials containing plenty OH groups may also create hydrogen bonds between molecules.
LIQUID SURFACE FILM THEORY:
The liquid-surface film theory attributes bonding to the presence of a thin liquid film which may be the
consequence of fusion or solution at the surface of the particle, induced by the energy of compression.
SOLID BRIDGES: The formation of solid bridges, also referred to as the diffusion theory of bonding,
occurs when two solids are mixed at their interface and accordingly to form a continuous solid phase.
HOT WELDING: Under the influence of applied pressure, an edge of the contact points between particles
undergoes a possible melting due to generation of heat in case of low melting point solids. Under
unloading of stress these melted point of contacts undergo re-solidification, forming a solid bridge
between the particles.](https://image.slidesharecdn.com/fullip1-151104153439-lva1-app6892/85/Industrial-Pharmacy-Notes-for-M-Pharmacy-19-320.jpg)




















![Patent
• Patents shall be granted for any inventions, whether products or processes, provided they are new,
involve an inventive step, & are capable of industrial application.
• Patents shall be granted in all fields of technology.
Trademark
• Defines what types of signs must be eligible for protection as trademarks.
• Service marks protected the same way.
Copyright
• Protection of computer programs as literary works & of compilations of data.
• The agreement says performers must also have the right to prevent unauthorized recording,
reproduction and broadcast of live performances (bootlegging) for no less than 50 years.
Industrial Designs
• Protection should be conferred on designs which are new or original.
• Exclusive rights can be exercised against acts for commercial purposes, including importation.
• The minimum term of protection is 10 years
Trade Secrets
• Undisclosed commercial information is to be protected against unfair commercial practices
• Secret data submitted for the approval of new chemical entities for pharmaceutical &
agrochemical products should be protected against unfair commercial use & disclosure by
governments.
Access to essential medicines
The most visible conflict has been over AIDS drugs in Africa. Despite the role that patents have played in
maintaining higher drug costs for public health programs across Africa, this controversy has not led to a
revision of TRIPs. Instead, an interpretive statement, the Doha Declaration, was issued in November 2001,
which indicated that TRIPs should not prevent states from dealing with public health crises. After
Doha, PhRMA, the United States and to a lesser extent other developed nations began working to
minimize the effect of the declaration.[7]
A 2003 agreement loosened the domestic market requirement, and allows developing countries to export
to other countries where there is a national health problem as long as drugs exported are not part of a
commercial or industrial policy.[8]
Drugs exported under such a regime may be packaged or colored
differently in order to prevent them from prejudicing markets in the developed world.
In 2003, the Bush administration also changed its position, concluding that generic treatments might in
fact be a component of an effective strategy to combat HIV. Bush created the PEPFAR program, which
received $15 billion from 2003–2007, and was reauthorized in 2008 for $48 billion over the next five
years. Despite wavering on the issue ofcompulsory licensing, PEPFAR began to distribute generic drugs
in 2004-5.
IMPLEMENTATION & IMPACT](https://image.slidesharecdn.com/fullip1-151104153439-lva1-app6892/85/Industrial-Pharmacy-Notes-for-M-Pharmacy-42-320.jpg)



























![INTRODUCTION
Pilot plant technique is defined as a part of the pharmaceutical industry where a lab scale process is
transformed into a viable product by the Development of liable practical procedure for manufacture of
dosage forms. The Scale-up is the art of designing of prototype using the data obtained from the pilot plant
model.
The Objective of Scale up Technique
To develop and formulate physically and chemically stable therapeutic dosage forms by optimizing
various parameters. To create a guidelines for production and process control. Raw materials handling
and its specifications requirements To identify the critical steps involved in the process. To develop a
master manufacturing formula. Pilot plant studies may be developed to establish the identical
examination of the formula to withstand batch scale. Infrastructure the related to scale up efforts in the
pilot plant: Production and process controls are evaluated, validated and finalized. Any Process
modification can be allowed To Evaluate and validate the developed product. To update the processing
equipment. Physical and mechanical Compatibility of the equipment with the formulation. Time and
cost factor. Need for current market strategies. To overcome the difficulties in small scale and create
large scale production.
Significance of Pilot Plant [3]
Standardization of formulae. Review of range of relevant processing equipments. Optimization and
control of production rate. Information on infrastructure of equipments during the scale up batches
physical space required. Identification of critical features to maintain quality of a product. Appropriate
records and reports to support GMP.
Pilot Plant Design for Tablets:
The primary responsibility of the pilot plant staff is to ensure that the newly formulated tablets developed
by product development personnel will prove to be efficiently, economically, and consistently
reproducible on a production scale. The design and construction of the pharmaceutical pilot plant for
tablet development should incorporate features necessary to facilitate maintenance and cleanliness. If
possible, it should be located on the ground floor to expedite the delivery and shipment of supplies. Each
stage considered carefully from experimental lab batch size to intermediate and large scale production.
Same process, same equipment but different performance when amount of material increased
significantly. May involve a major process change that utilizes techniques and equipment that were
either unavailable or unsuitable on a lab scale.
Stages of Production of Tablets
Material handling Dry blending Granulation Drying Reduction of particle size Blending Direct
compression Slugging (dry granulation)
Material Handling System
In the laboratory, materials are simply scooped or poured by hand, but in intermediate- or large-scale
operations, handling of this materials often become necessary. If a system is used to transfer materials for
more than one product steps must be taken to prevent cross contamination. Any material handling system
must deliver the accurate amount of the ingredient to the formulation. The More sophisticated methods of
handling materials arevacuum loading systems, metering pumps, screw feed system. The types of the
system selected depend on the nature of the materials, e.g., density and static change.](https://image.slidesharecdn.com/fullip1-151104153439-lva1-app6892/85/Industrial-Pharmacy-Notes-for-M-Pharmacy-79-320.jpg)






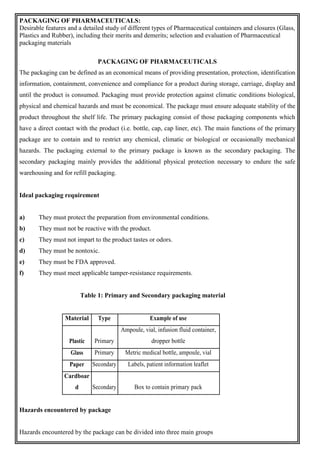








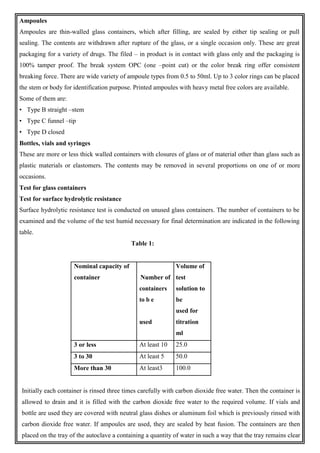




















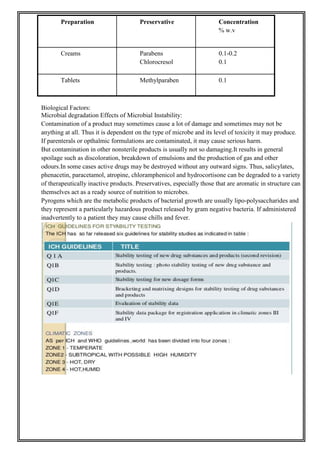





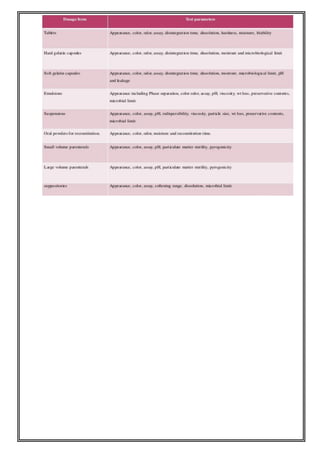











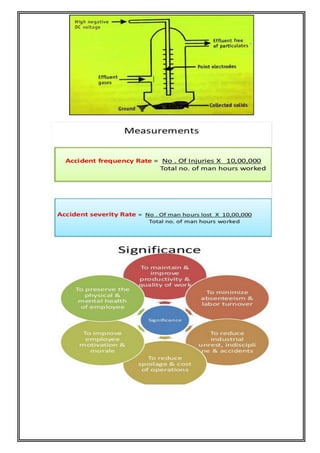


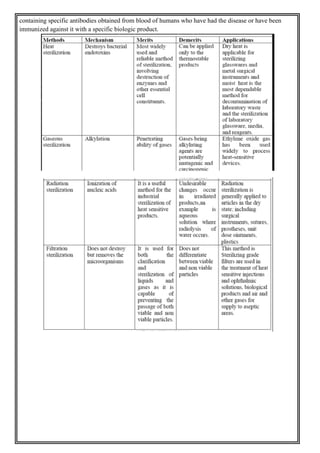
This document provides an overview of preformulation studies for developing drug formulations. It discusses the importance of studying the physical and chemical properties of drug substances alone and when combined with excipients. The summary is: 1) Preformulation studies investigate the physical and chemical properties of drugs and how they interact with excipients. This helps formulators develop stable, bioavailable drug products. 2) Key properties studied include solubility, particle size, solid state properties, and how the drug behaves under different conditions like temperature, light and pH. 3) The results of preformulation studies guide formulation development and ensure the final product has the desired quality, performance and safety.



















![Sustained release dosage form [srdf]](https://cdn.slidesharecdn.com/ss_thumbnails/sustainedreleasedosageformsrdf-160516130442-thumbnail.jpg?width=640&height=640&fit=bounds)













































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)

















