Downloaded 348 times



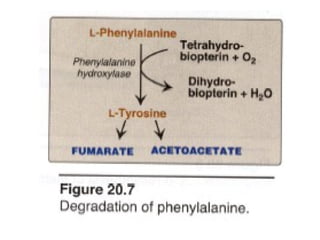
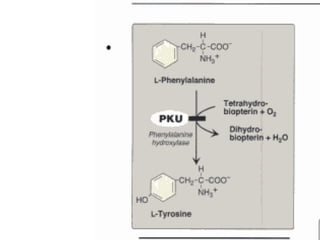
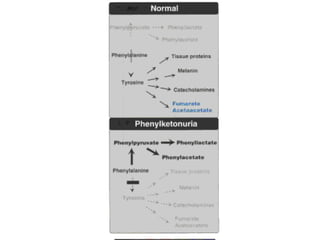
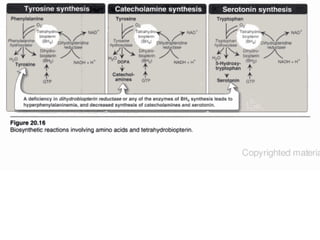









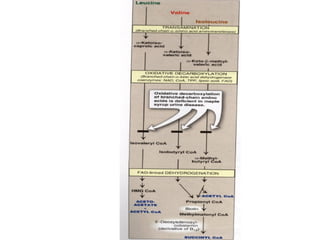






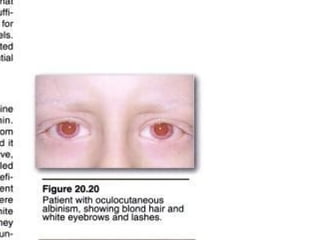






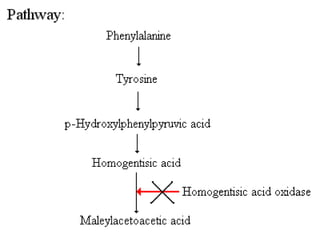





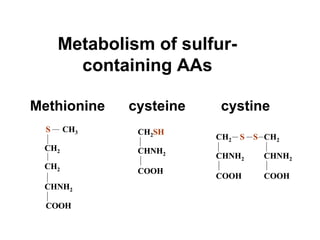

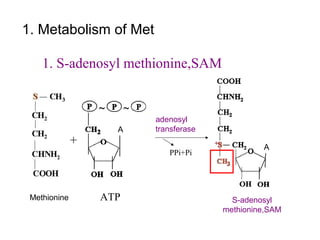
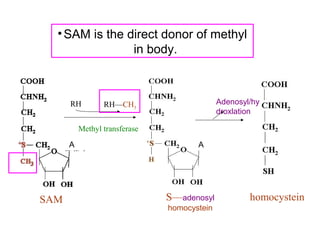
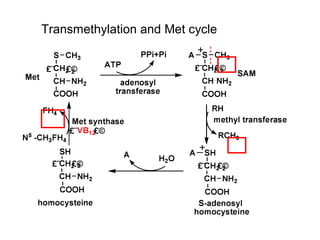
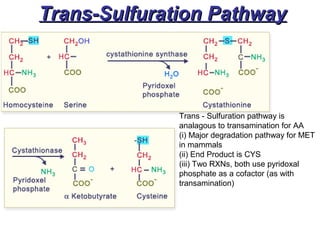
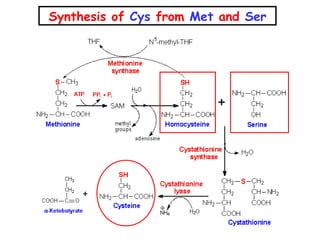

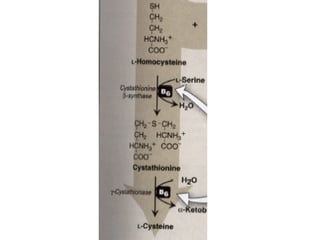
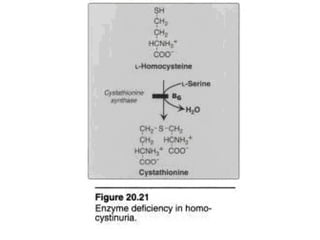




This document discusses several inherited metabolic disorders involving amino acid metabolism. Phenylketonuria is described as the most common disorder, caused by a deficiency of the enzyme phenylalanine hydroxylase, leading to toxic accumulation of phenylalanine. Maple syrup urine disease results from a defect in the enzyme branched-chain keto acid dehydrogenase, causing a buildup of leucine, isoleucine, and valine breakdown products. Homocystinuria is caused by a cystathionine beta-synthase deficiency, preventing the breakdown of homocysteine. These disorders are typically detected via newborn screening and require dietary modifications and supplements to prevent associated complications.



































































