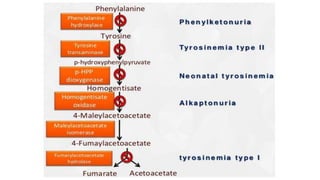
Tyrosinosis is a genetic disorder caused by enzyme deficiencies that disrupt normal metabolism, leading to toxic accumulation of tyrosine and its byproducts, resulting in serious health issues. There are three types of tyrosinemia, each linked to different enzyme deficiencies, with type I being the most severe, potentially causing liver failure or cancer. Diagnosis includes elevated levels of tyrosine in the blood and urine, and treatment involves the use of nitisinone and dietary management to control tyrosine levels.