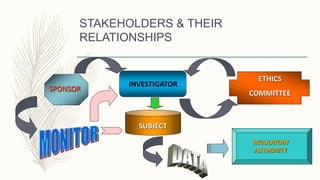
Good Clinical Practice (GCP) guidelines provide standards for conducting clinical trials involving human subjects. The history of GCP includes events like the Nuremberg Code (1949), Declaration of Helsinki (1964), and ICH guidelines (1990-1997) that were developed in response to ethical issues in clinical research. The ICH GCP guideline has 8 sections covering topics like investigator responsibilities, informed consent, and essential trial documents. GCP aims to protect subject rights and safety while ensuring reliable trial data.