Downloaded 56 times


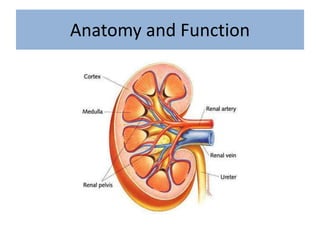

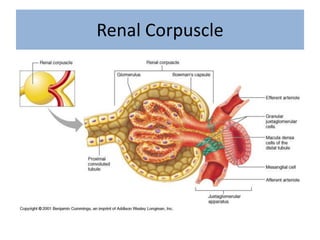

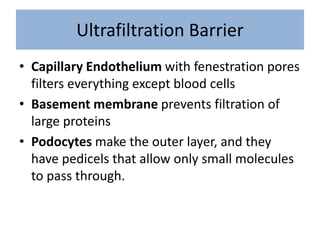

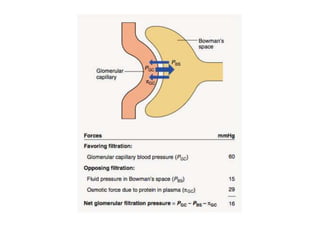











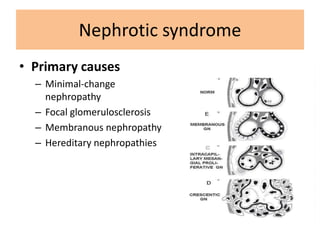


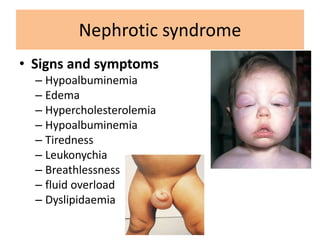





































1. Glomerular diseases involve inflammation or disorders of the glomerulus in the kidney. Common glomerular diseases include glomerulonephritis and glomerulopathy. 2. Proteinuria and hematuria are common signs of glomerular disease. Persistent proteinuria or hematuria should be investigated further. Causes can include infections, autoimmune diseases, genetic factors, metabolic disorders and other systemic illnesses. 3. Glomerular diseases are classified based on factors such as location of involvement in the glomerulus (focal vs diffuse) and whether the primary involvement is of the glomerulus or a secondary manifestation of another disease. Immune mechanisms underlie many primary and secondary glomerular































![Nephrotic syndrome [full]](https://cdn.slidesharecdn.com/ss_thumbnails/nephroticsyndromefull-161026190255-thumbnail.jpg?width=640&height=640&fit=bounds)


















































