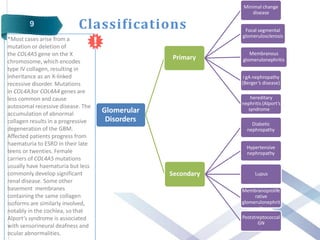
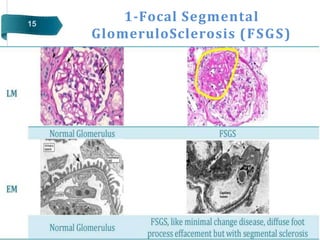
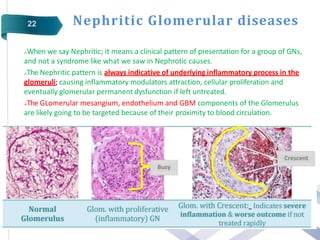
Glomerular diseases can be classified as either primary (intrinsic renal pathology) or secondary (to a systemic disease). They present clinically with nephritic syndrome (hematuria, hypertension, azotemia) or nephrotic syndrome (proteinuria, edema, hypoalbuminemia, hyperlipidemia). The pathophysiology depends on which part of the glomerular structure is affected - podocyte damage leads to nephrotic syndrome with proteinuria alone, while damage to endothelial cells, GBM or mesangial cells causes nephritic syndrome with hematuria and proteinuria. Common primary glomerular diseases include minimal change disease, focal segmental glomerulosclerosis, membranous glomerulone































































![Nephrotic syndrome [full]](https://cdn.slidesharecdn.com/ss_thumbnails/nephroticsyndromefull-161026190255-thumbnail.jpg?width=640&height=640&fit=bounds)






























