1) Glomerular diseases involve either nephrotic syndrome characterized by massive proteinuria or nephritic syndrome characterized by hematuria and inflammation.
2) Workup includes labs, urinalysis, protein quantification, serologies, and often kidney biopsy.
3) Common causes discussed include minimal change disease, focal segmental glomerulosclerosis, membranous nephropathy, acute poststreptococcal glomerulonephritis, and membranoproliferative glomerulonephritis.


![Workup
• Basic laboratory testing (including plasma or serum creatinine, albumin,
electrolytes, calcium, phosphorus, and complete blood count)
• urinalysis of sediment (critical in establishing the presence of most of the nephritic
syndromes; red blood cell [RBC] casts or dysmorphic RBCs are hallmarks)
• estimation of protein excretion (spot protein/creatinine ratio or 24-hour urine
collection)
• serologic testing for disorders that cause glomerular disease (e.g., lupus and
hepatitis serologies, antistreptolysin O [ASO], cryogobulins, antinuclear antibody,
ANCA, anti-GBM, and protein electrophoresis)
• measurement of serum complement levels
• kidney biopsy (generally required to document the underlying pathology and to
identify the type of glomerular disease)
– Positive ANCA or anti-GBM tests in the context of nephritic syndrome may be sufficient to
make the diagnosis without the need for a biopsy, depending on the clinical status and kidney
function.
– If membranous nephropathy is seen on biopsy, then molecular and antibody studies should be
considered.](https://image.slidesharecdn.com/glomerulonephropathypgy1-230217040357-9a1bfec1/85/Glomerulonephropathy-PGY-1-pptx-4-320.jpg)
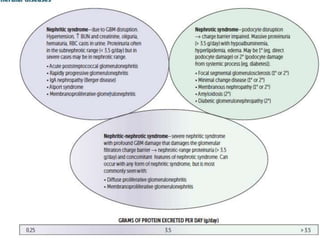
![Nephrotic Syndrome
• Nephrotic syndrome NephrOtic syndrome—massive prOteinuria (>
3.5 g/day) with hypoalbuminemia, resulting edema, hyperlipidemia.
Frothy urine with fatty casts.
• Disruption of glomerular filtration charge barrier may be primary
(eg, direct sclerosis of podocytes) or secondary (systemic process
[eg, diabetes] secondarily damages podocytes).
• Severe nephritic syndrome may present with nephrotic syndrome
features (nephritic-nephrotic syndrome) if damage to GBM is severe
enough to damage charge barrier.
• Associated with hypercoagulable state due to antithrombin (AT) III
loss in urine and •
risk of infection (loss of immunoglobulins in urine
and soft tissue compromise by edema).](https://image.slidesharecdn.com/glomerulonephropathypgy1-230217040357-9a1bfec1/85/Glomerulonephropathy-PGY-1-pptx-5-320.jpg)





























































