
Downloaded 32 times






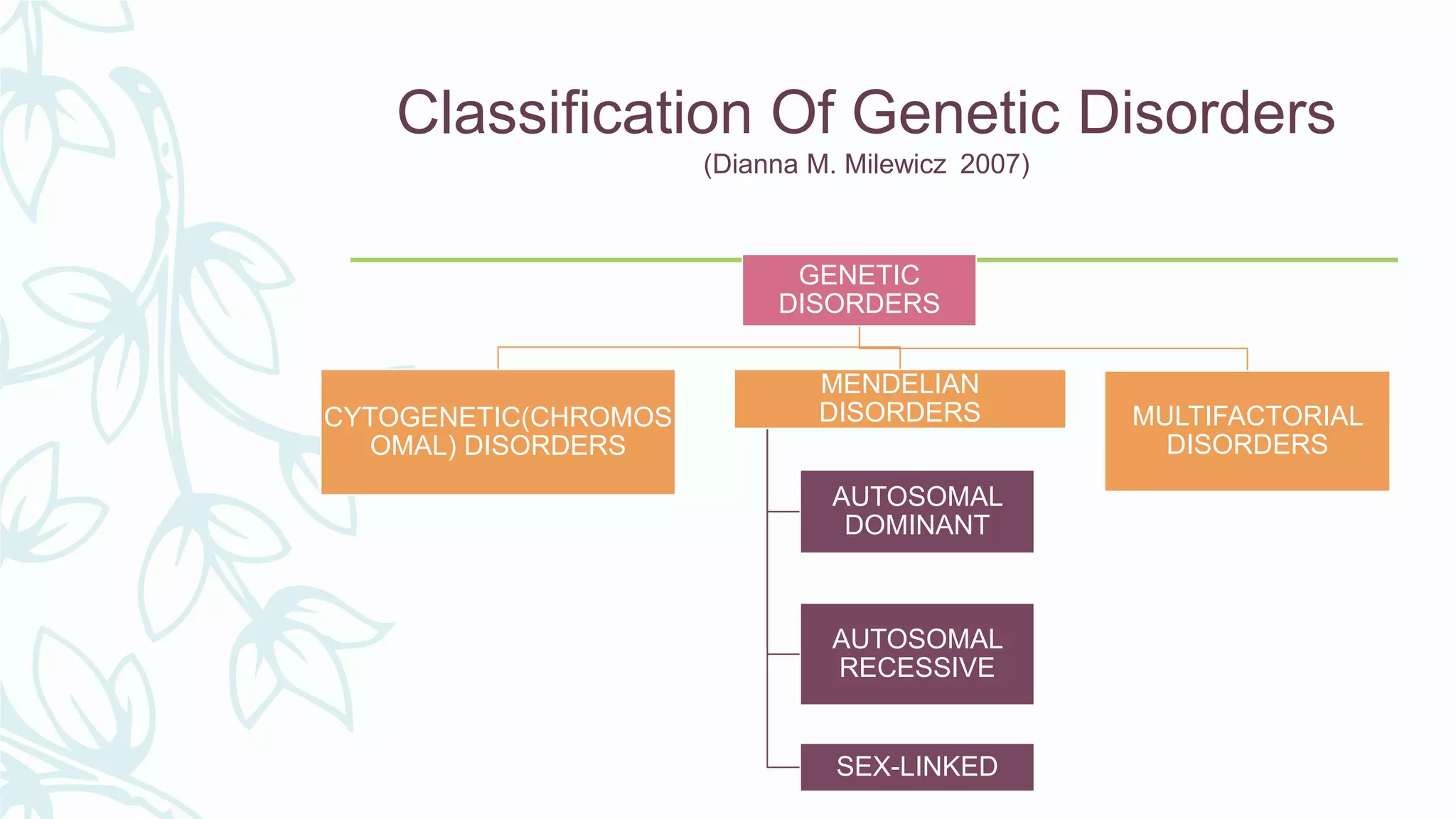
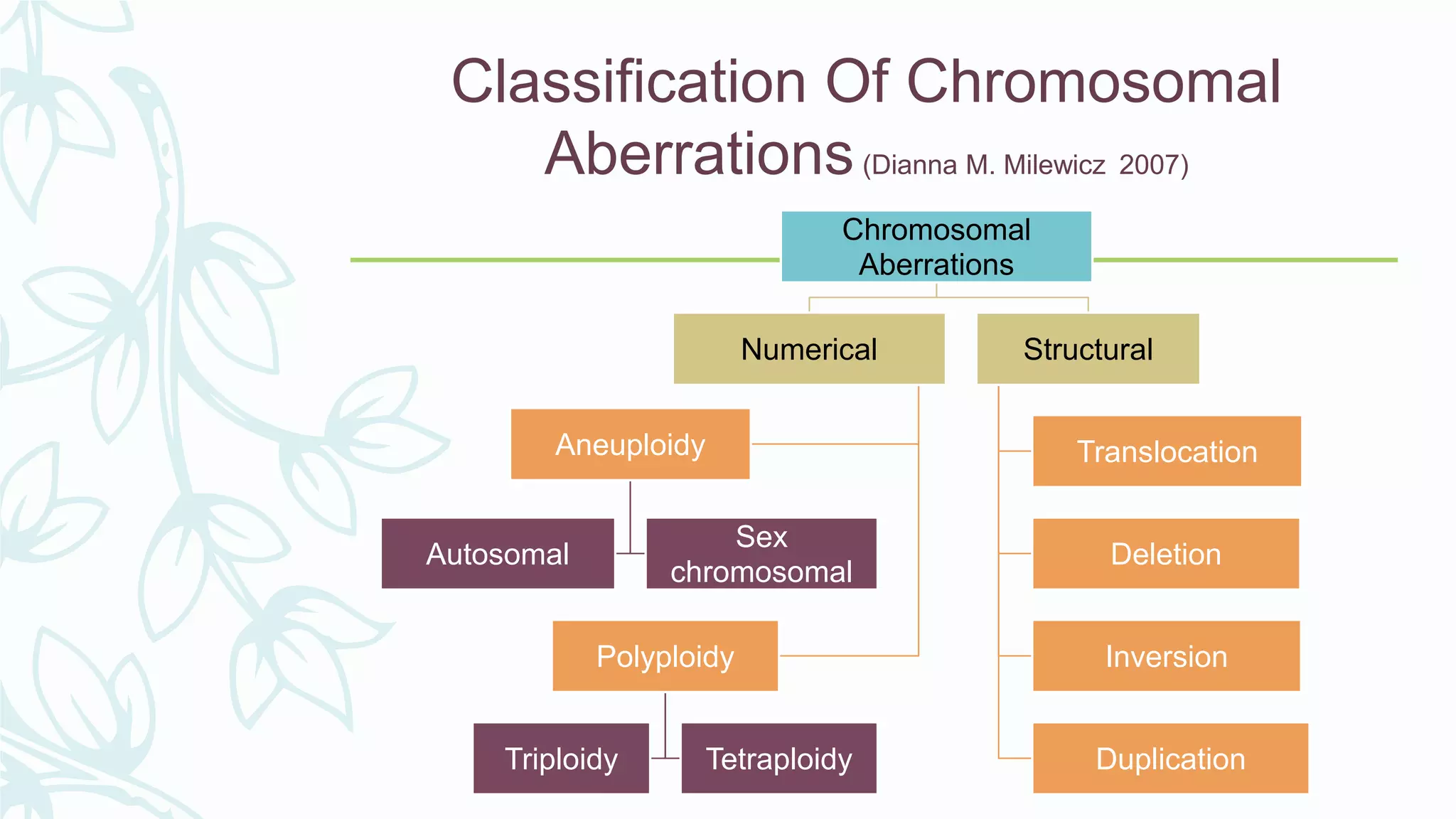








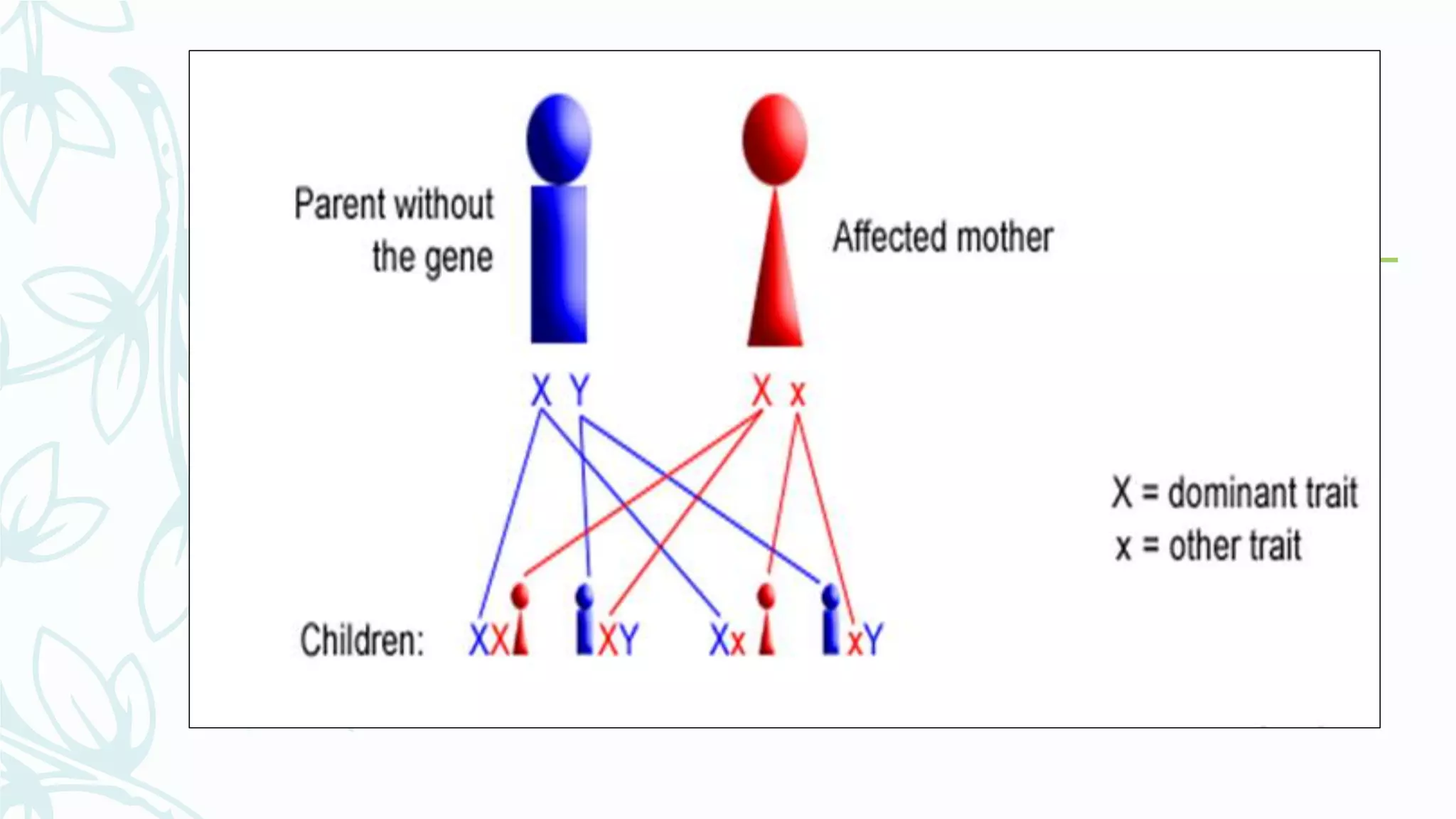



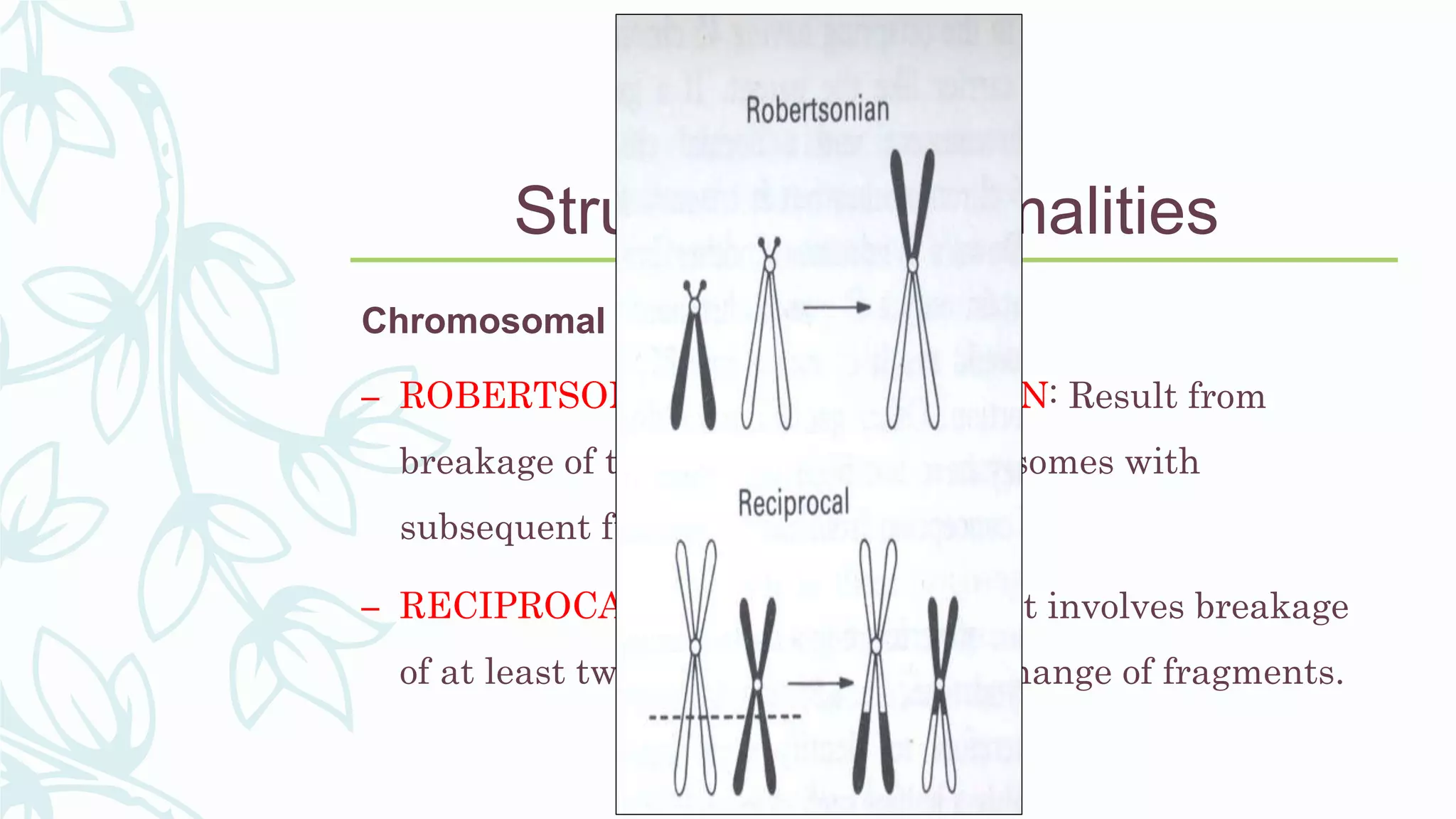













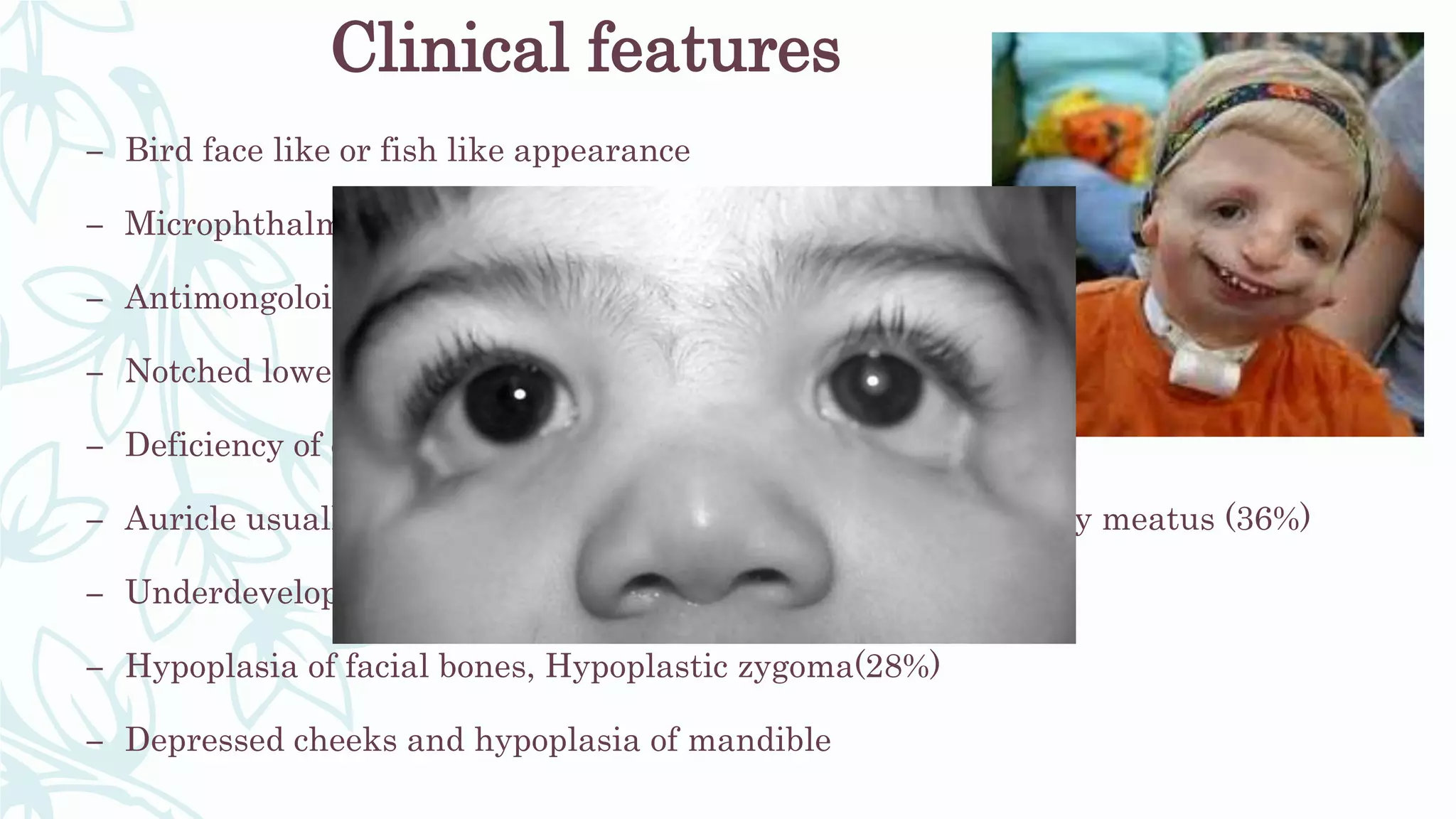



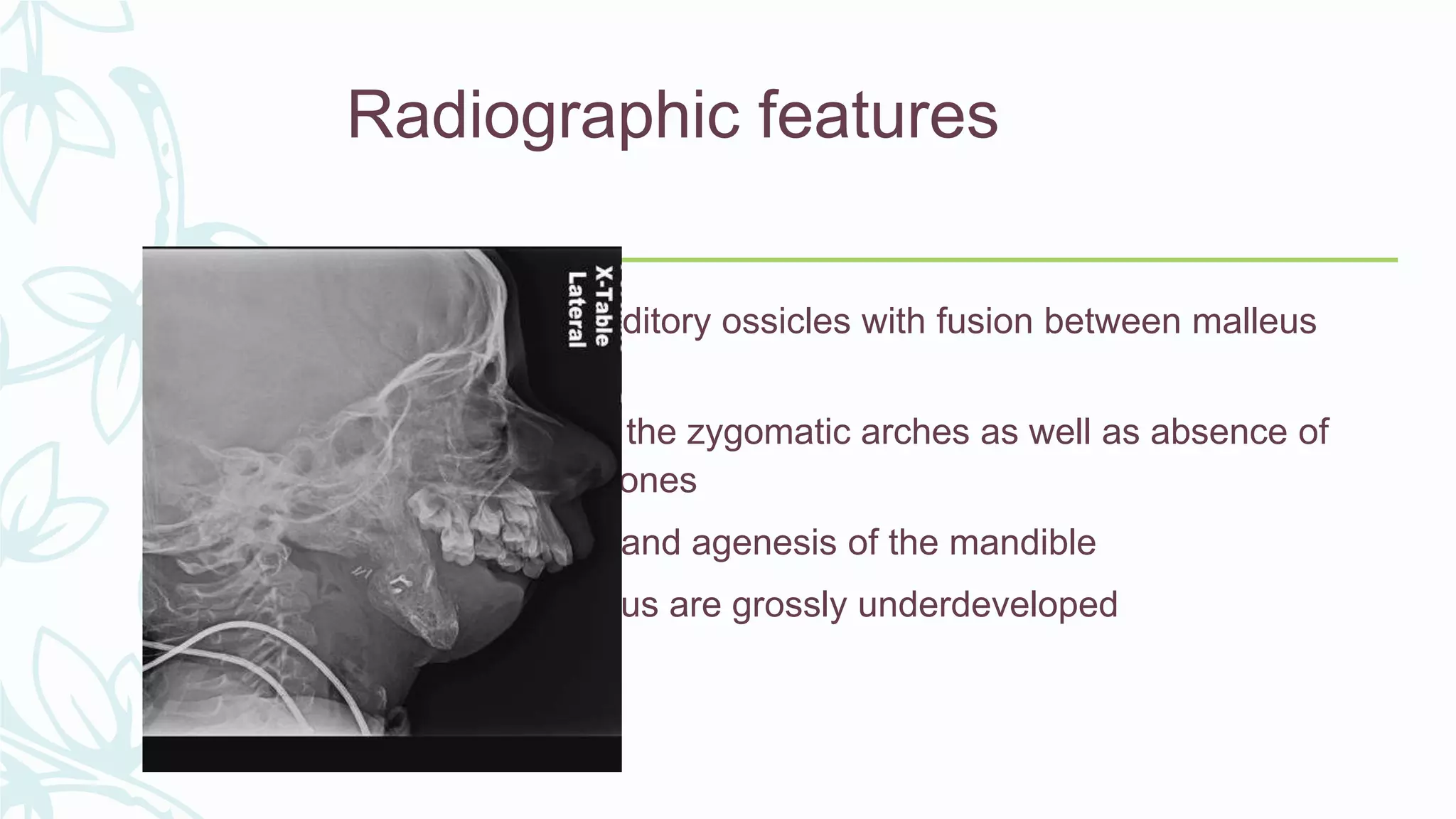






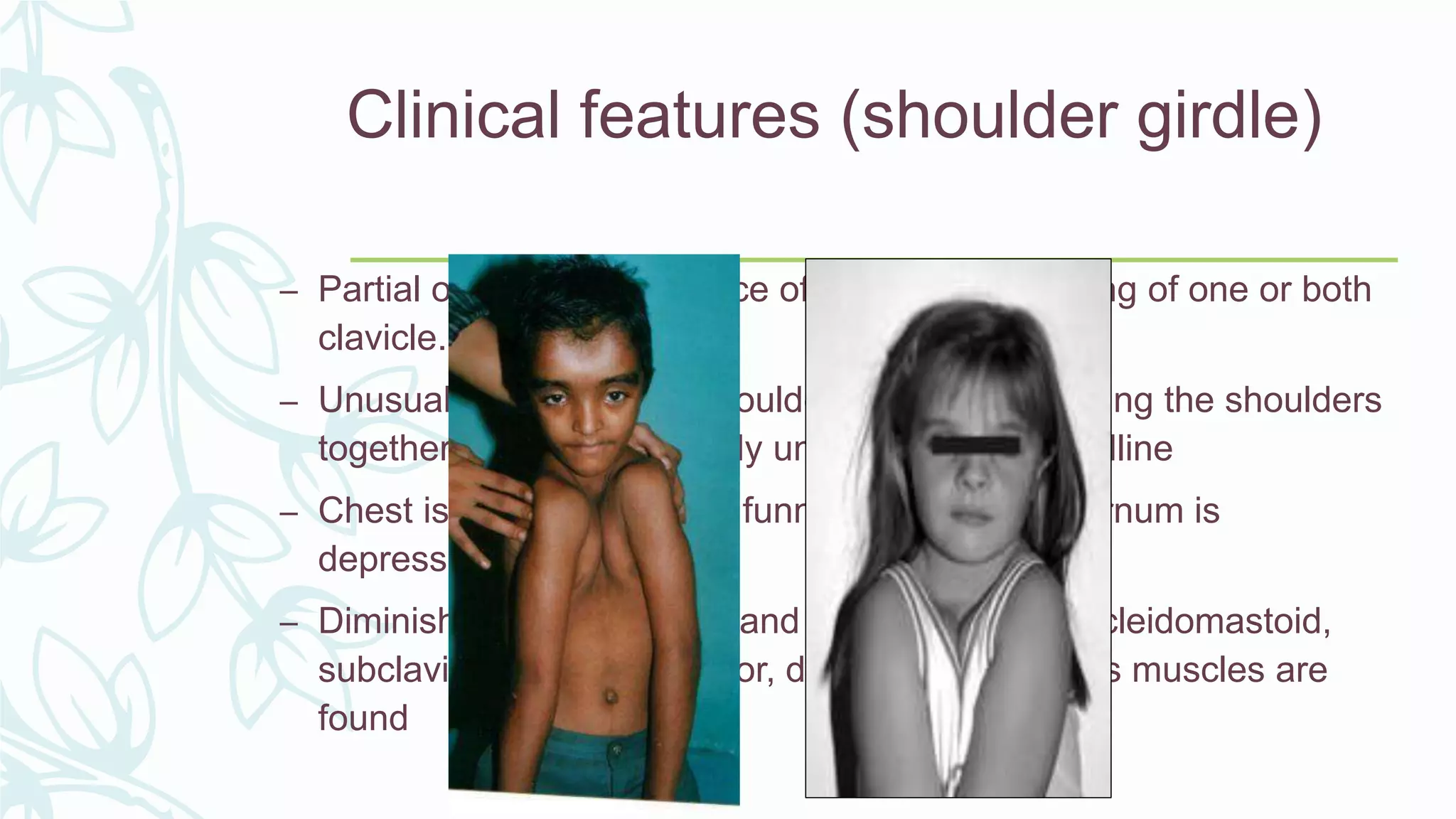





















































































































The document provides an extensive overview of genetic and chromosomal disorders in children, outlining definitions, classifications, and specific conditions associated with genetic aberrations. It discusses inheritance patterns, types of chromosomal abnormalities, and details various genetic disorders like achondroplasia and Marfan's syndrome, with emphasis on clinical features and treatment options. The importance of recognizing these disorders by dental professionals is highlighted, as they play a key role in identifying unrecognized genetic issues in patients.













































































