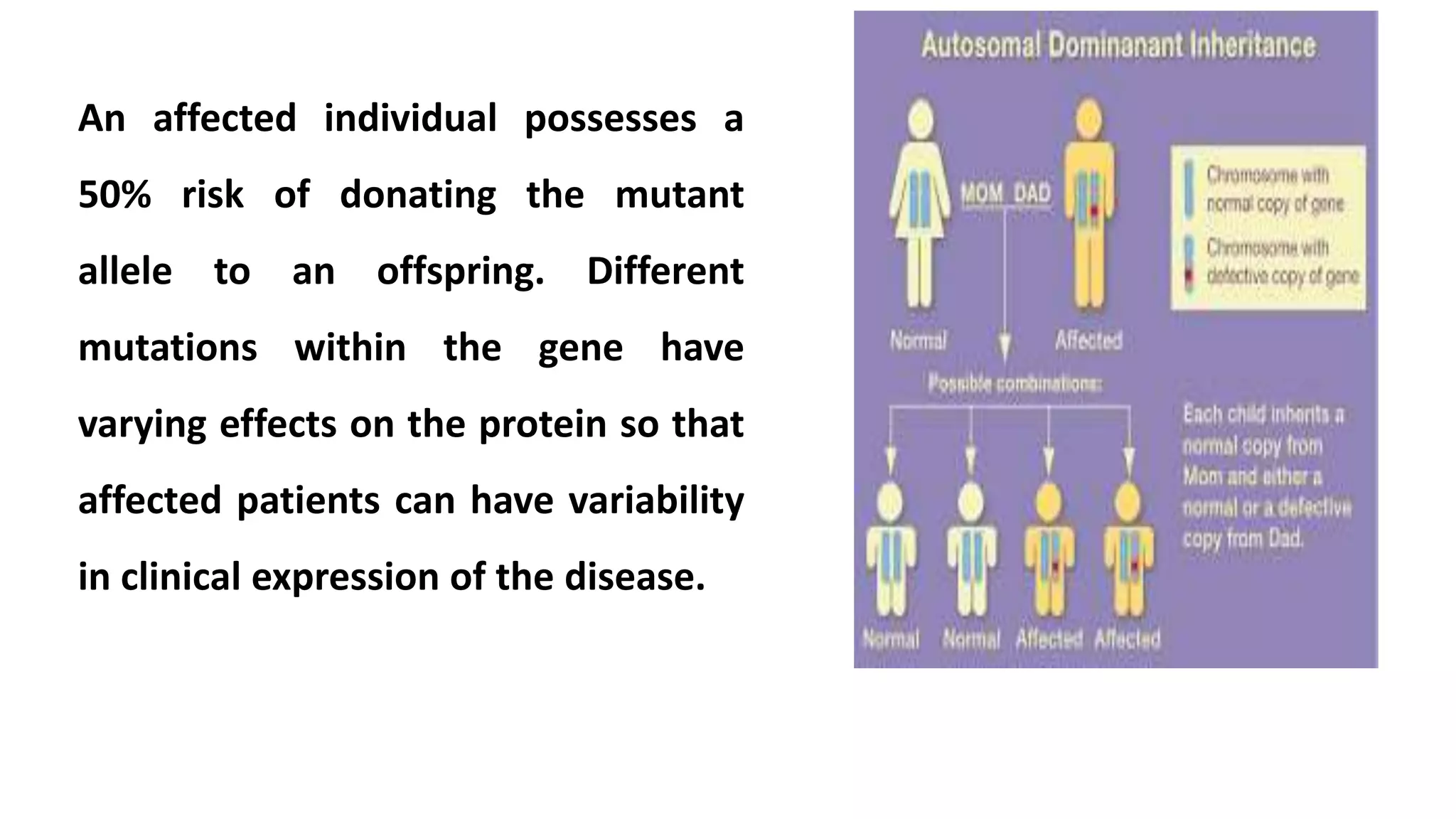
1. Autosomal dominant diseases are inherited disorders caused by mutations in a single gene located on chromosomes 1-22. An affected individual has a 50% chance of passing on the mutant allele to their offspring. Variable expression is common, even within families, due to modifier genes and environmental factors.
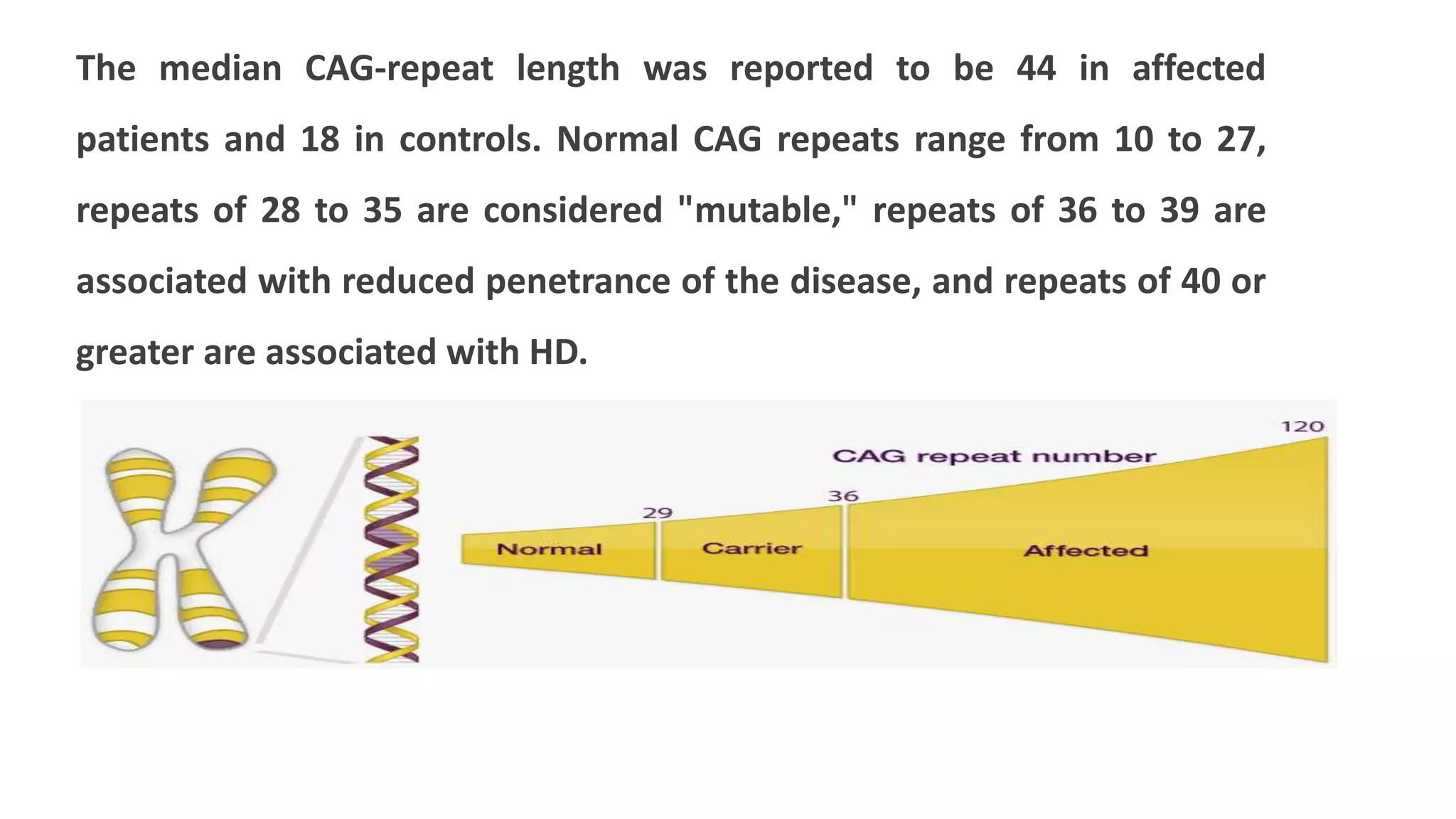
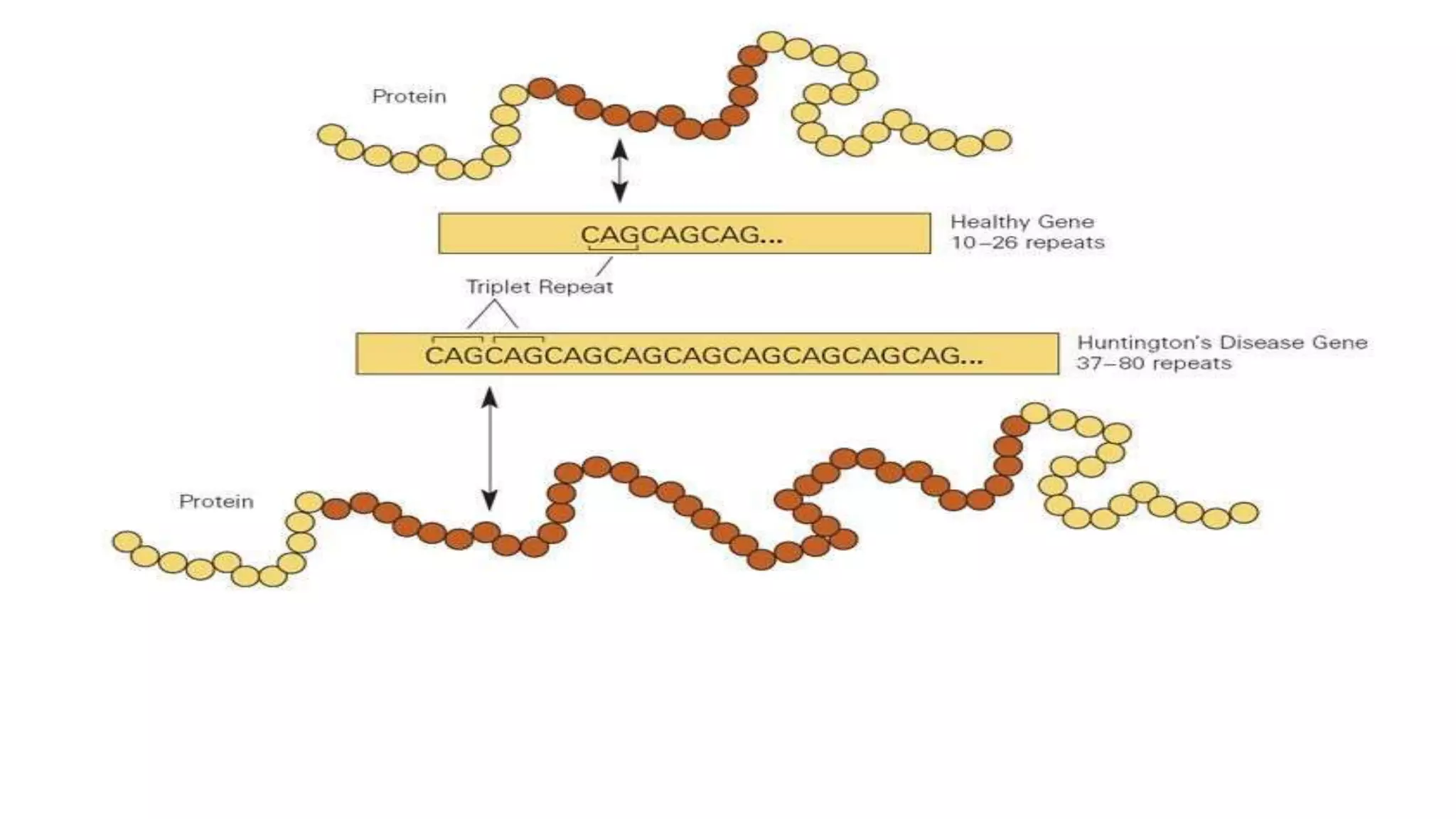
2. Achondroplasia is caused by mutations in the FGFR3 gene leading to dwarfism. Charcot-Marie-Tooth disease type 1A is caused by duplications in the PMP22 gene resulting in peripheral neuropathy. Huntington's disease causes chorea and dementia and is associated with CAG repeat expansions in the HTT gene.




























































































![Genetic Basis & Clinical Features of Achondroplasia [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/geneticbasisclinicalfeaturesofachondroplasiaautosaved-240504095205-862c43d1-thumbnail.jpg?width=640&height=640&fit=bounds)














































