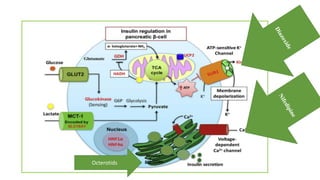
Congenital hyperinsulinism is a rare genetic disorder characterized by inappropriate insulin secretion that causes recurrent episodes of low blood sugar (hypoglycemia). It has an incidence of 1 in 50,000 live births. The condition is caused by mutations in genes that regulate insulin secretion from pancreatic beta cells. Treatment aims to maintain blood glucose levels above 63 mg/dl to prevent neurological harm. Management options include oral medications, intravenous glucose, glucagon injections, octreotide, surgery, and living with frequent feedings. Delayed diagnosis and treatment can lead to long-term neurological issues in over half of patients.
















![• Results
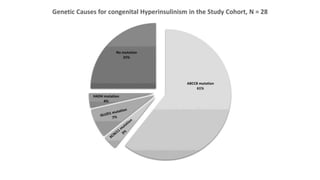
• ABCC8 mutation was found in (61%) of cases who underwent genetic testing
(17/28).
• Five cases with homozygous biparental ABCC8 mutation responded to
combined diazoxide and octreotide without needing surgery.
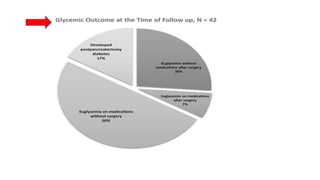
• Seven out of twenty-one patients who had pancreatectomy (33%) developed
diabetes after a median period of 4.8 (range:1–10) years following surgery.
• 55%e of our patients had neurodevelopmental impairment at follow-up.
• Logistic regression analysis has shown that delayed referral to tertiary centre
for more than 8 days, delayed diagnosis of CHI for more than 14 days and
hospital admission for more than 30 days, are significant predictors of
unfavorable neurological sequelae in CHI; (OR = 12.7 [2.56], p = 0.001),
(OR = 12.7 [2.9–56], p = 0.001), and (OR = 3.8 [0.14.5], p = 0.043),
respectively.](https://image.slidesharecdn.com/congenitalhyperinsuliism-221116230712-692155f2/85/Congenital-hyperinsuliism-pptx-17-320.jpg)






















































![Neonatal dm [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/neonataldmautosaved-200921092545-thumbnail.jpg?width=640&height=640&fit=bounds)









![Monkeypox [Autosaved].ppt](https://cdn.slidesharecdn.com/ss_thumbnails/monkeypoxautosaved-221001120403-0c64b98c-thumbnail.jpg?width=640&height=640&fit=bounds)




































