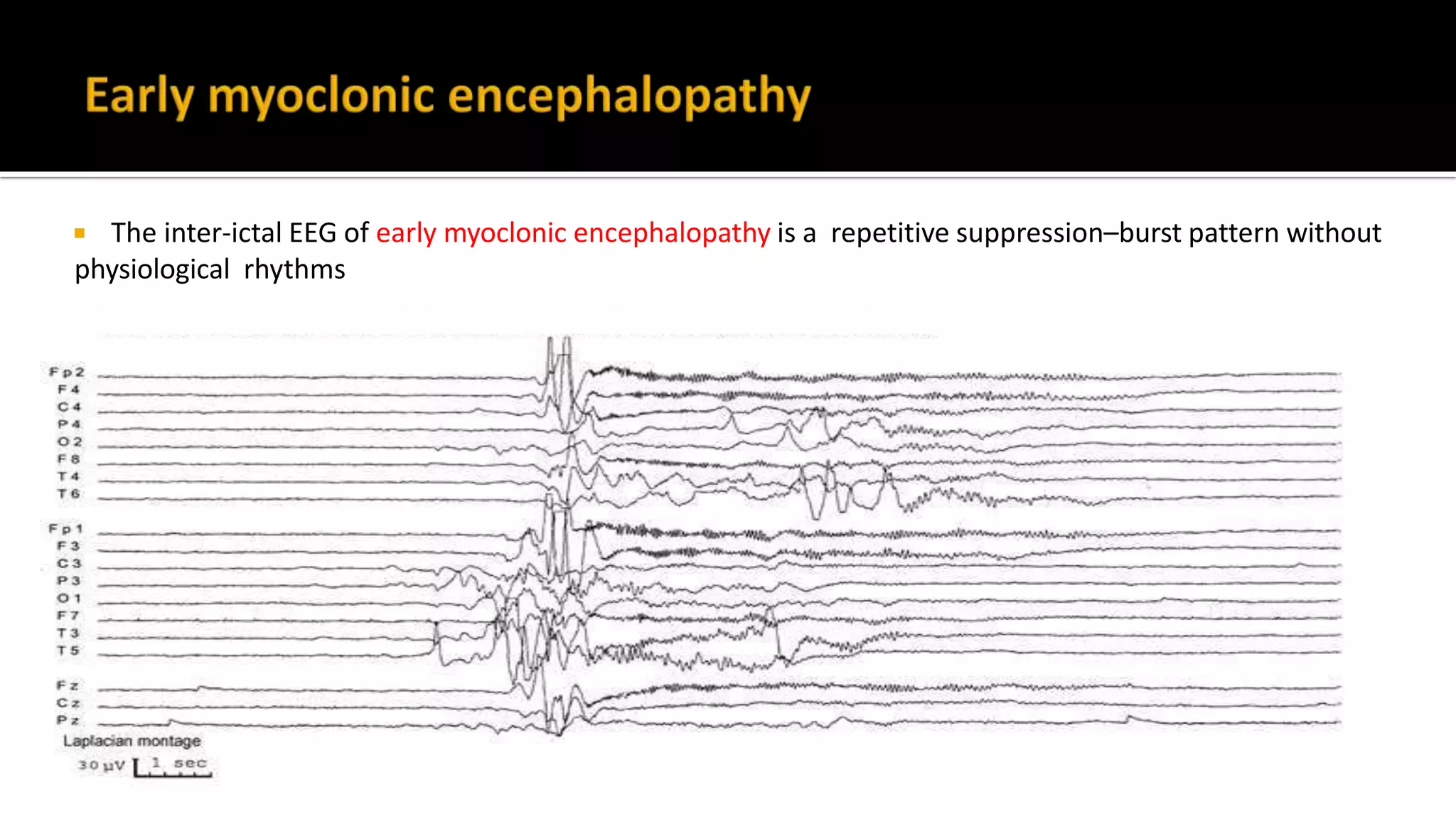
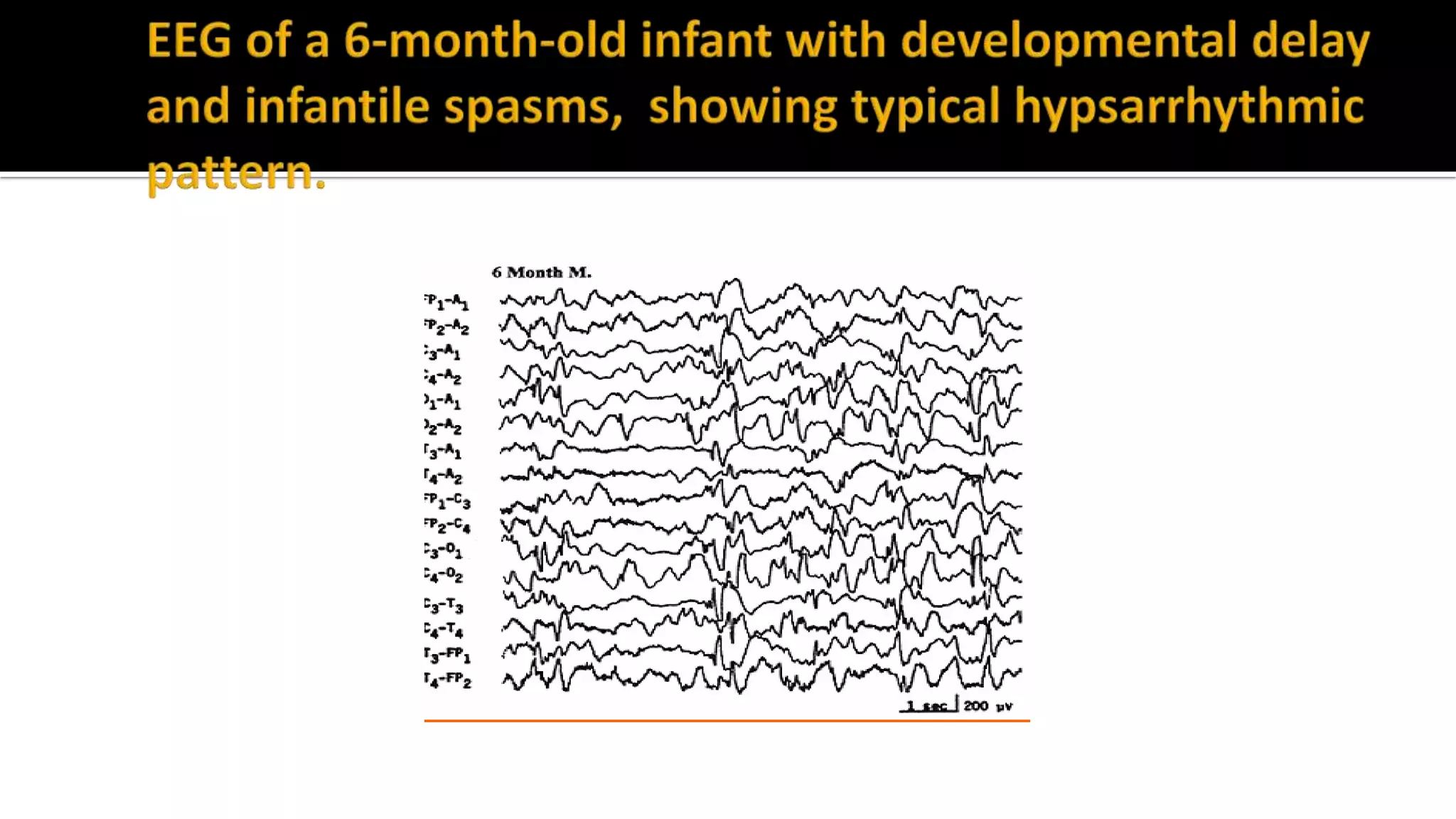
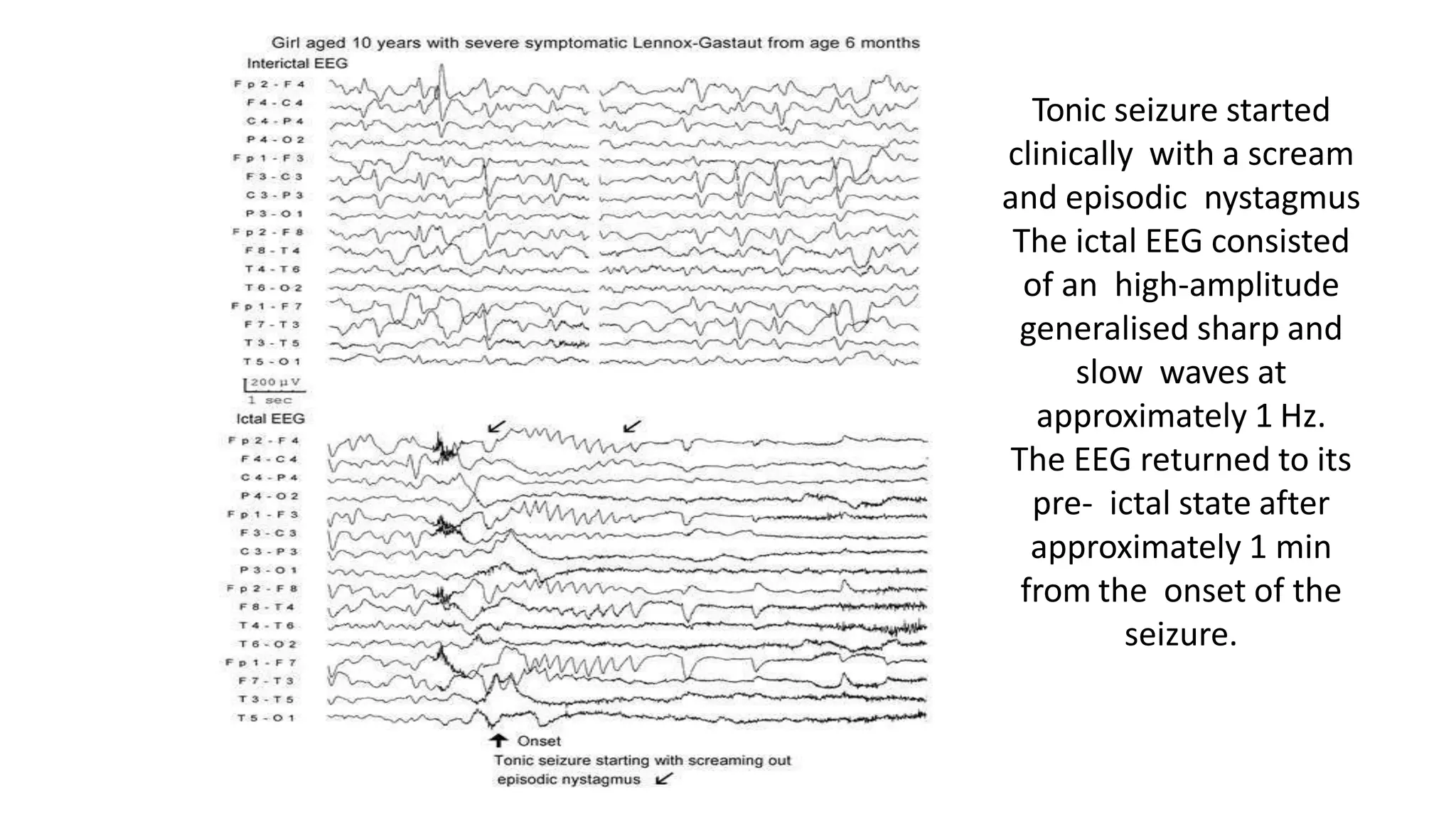
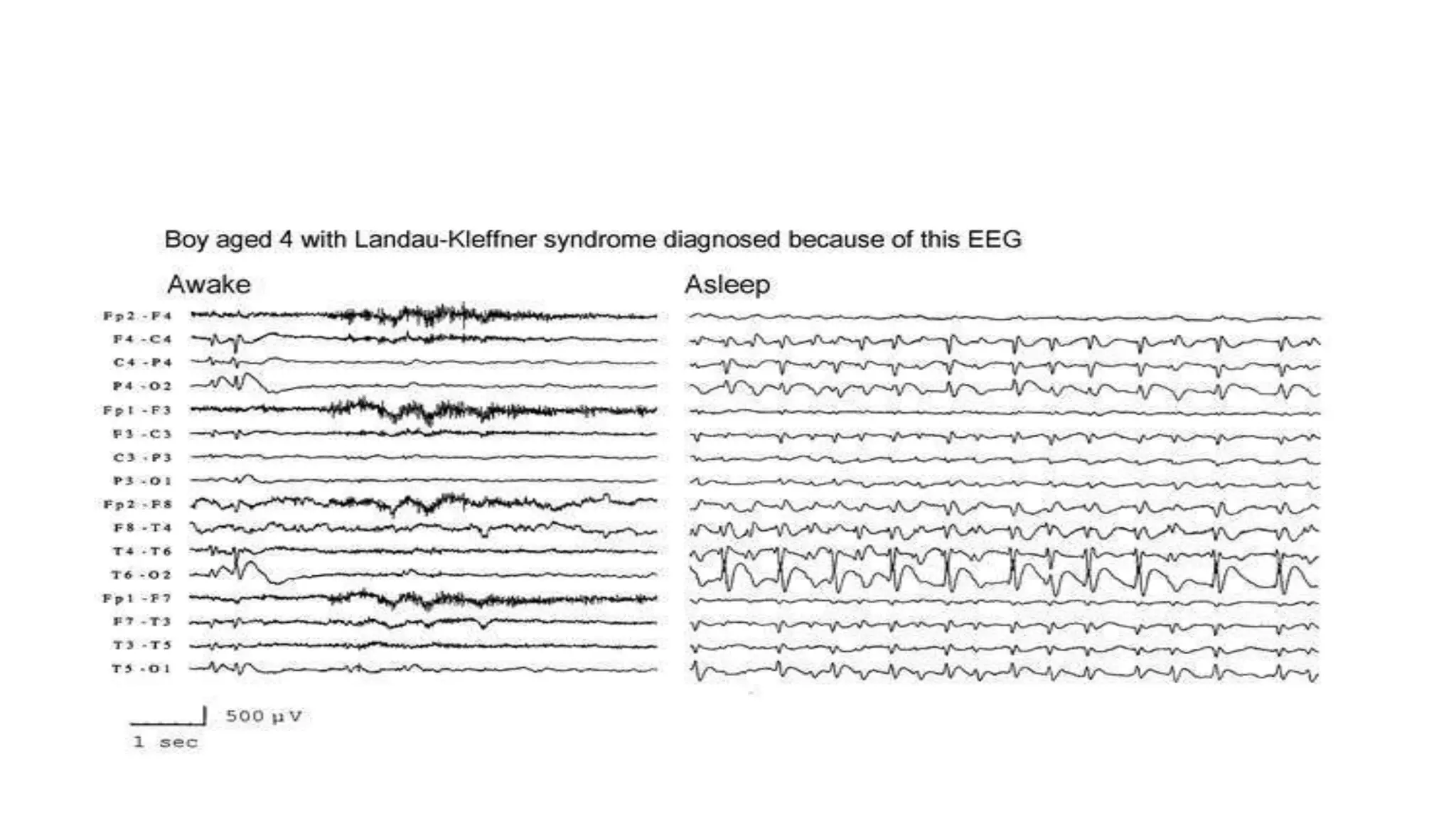
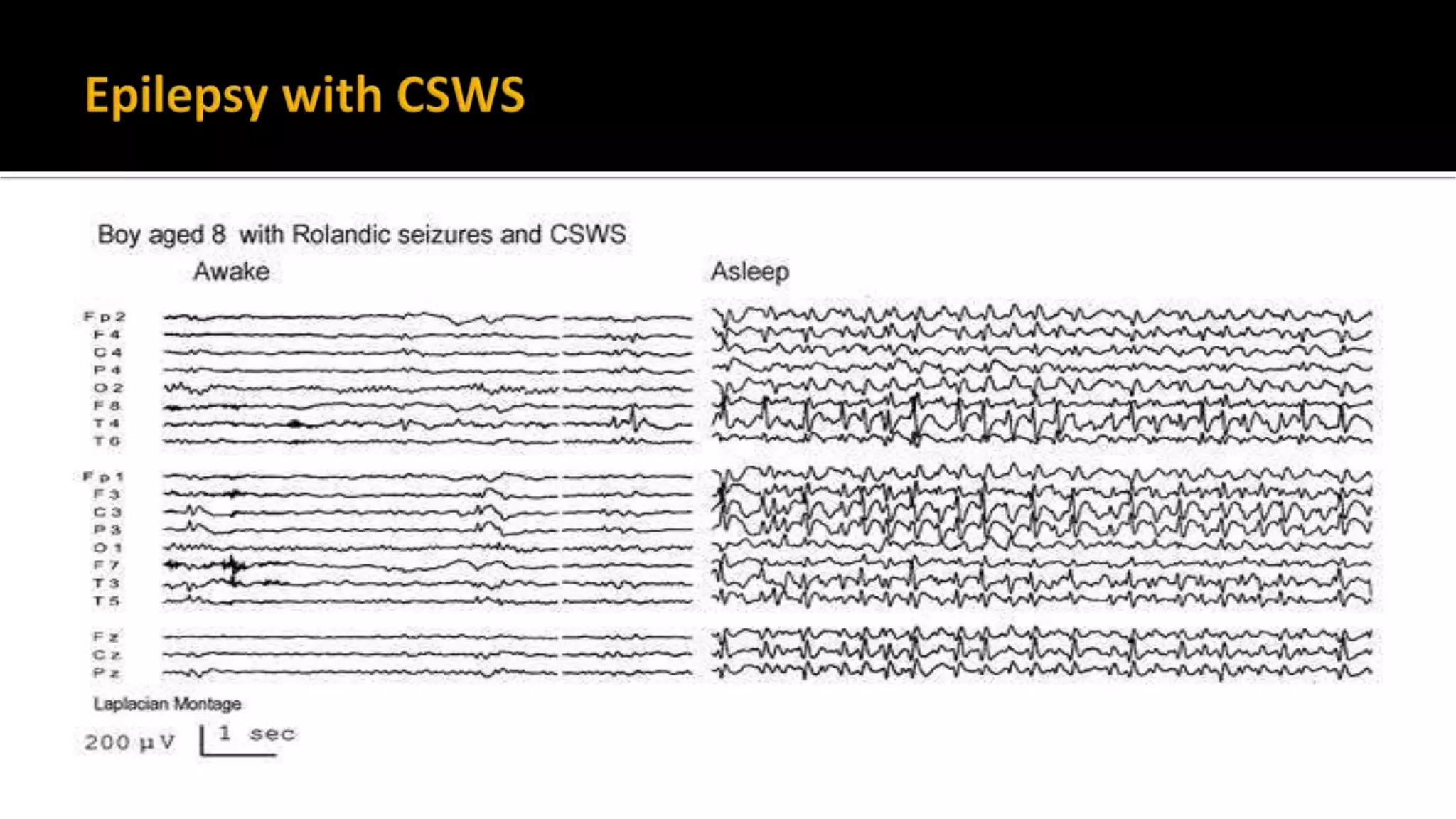
The document discusses epileptic encephalopathy syndromes that occur in neonates, infants, and children. It describes the key features of early myoclonic encephalopathy, West syndrome, Lennox-Gastaut syndrome, Landau-Kleffner syndrome, and continuous spike-wave during sleep. The syndromes are characterized by seizures, developmental regression or impairment, and specific electrographic findings on EEG. Prognosis varies between syndromes but is often poor, with cognitive and neurological deficits. Treatment involves antiepileptic drugs, corticosteroids, ketogenic diet, or surgery in refractory cases.




























































































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































