Downloaded 49 times





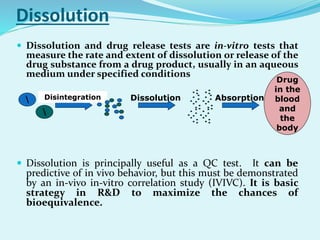
































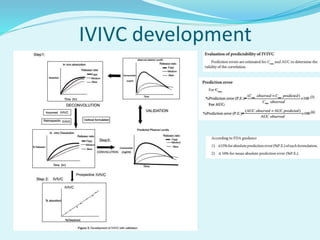






The document outlines the processes involved in drug discovery and pharmaceutical product development, including drug dissolution, disintegration testing, and the importance of in vitro and in vivo correlation (IVIVC). It details regulatory requirements, various dosage forms, and factors affecting drug dissolution, as well as the significance of comparative dissolution studies and the use of different dissolution apparatuses. Additionally, the document explores correlation levels in IVIVC which assist in optimizing formulations and reducing regulatory burdens.



































































