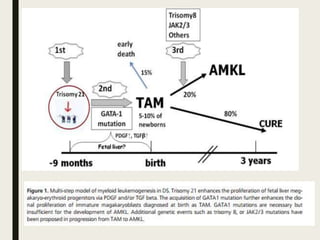
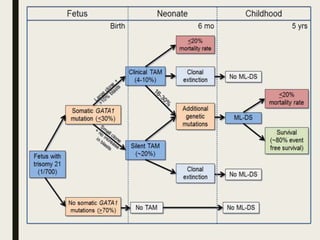
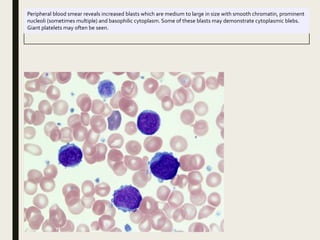
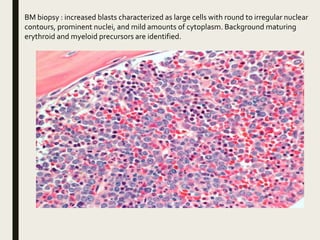
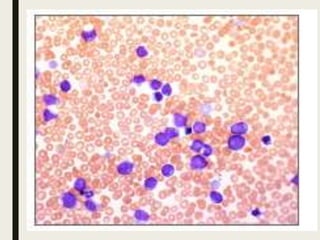
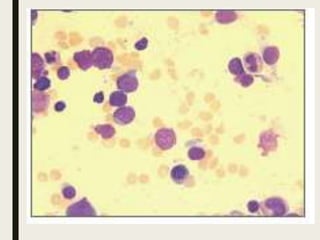
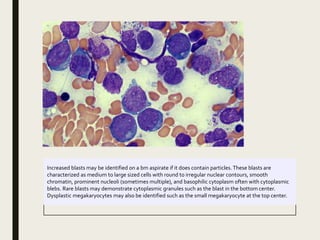
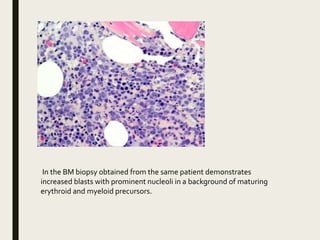
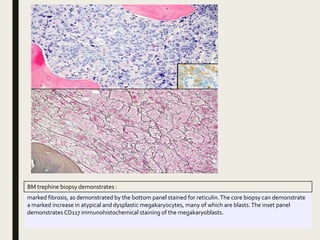
Down syndrome is a genetic disorder caused by trisomy 21 and is associated with an increased risk of certain leukemias. Transient abnormal myelopoiesis (TAM) occurs in 10% of newborns with Down syndrome and involves increased peripheral blood blasts. It typically resolves spontaneously within 3 months. Acute myeloid leukemia (AML) develops in 20-30% of children with a history of TAM and usually occurs 1-3 years later. AML in Down syndrome most commonly involves megakaryoblasts and is associated with mutations in the GATA1 gene. While chemotherapy is needed to treat AML, reduced intensity protocols can be used due to greater toxicity risks in Down syndrome patients.
















































































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)







![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)