This document provides an overview of bleeding disorders and their evaluation. It discusses:
1) The normal processes of hemostasis involving platelets, coagulation factors, and endothelium.
2) Classification of bleeding disorders as issues with blood vessels, platelets, or coagulation factors.
3) A stepwise diagnostic approach to evaluating bleeding disorders, beginning with patient history, clinical examination findings, and laboratory investigations.
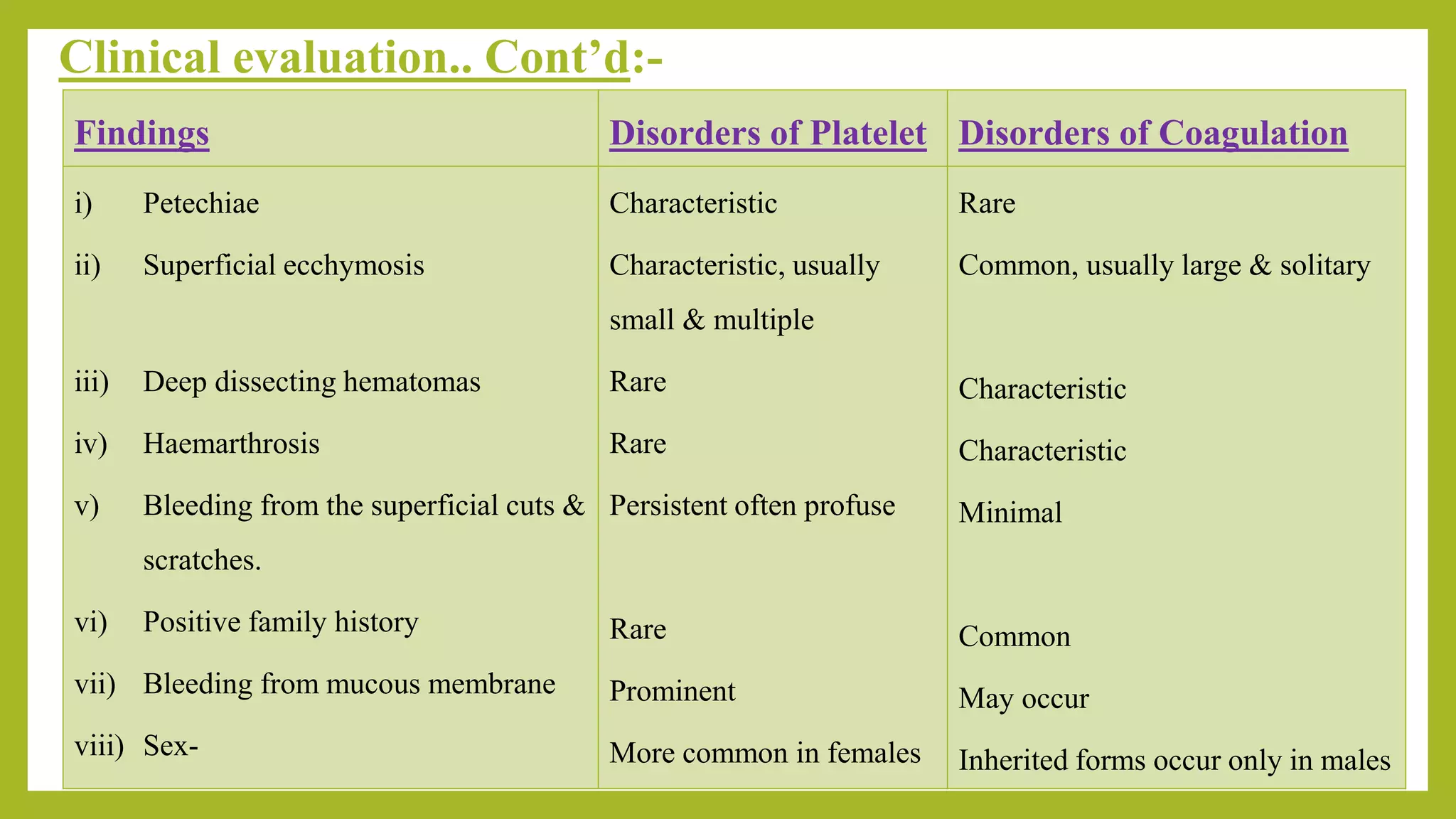
4) Details on using clinical features like bleeding pattern and location, family history, and physical exam findings to help differentiate between platelet disorders and coagulation factor deficiencies.