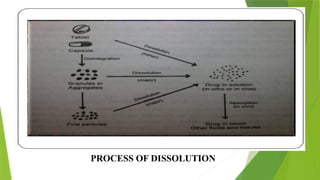
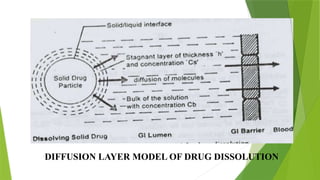
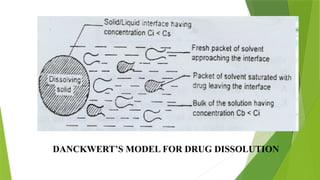
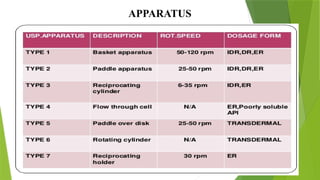
The document discusses the concept of dissolution, defining it as the process of a solid substance solubilizing in a solvent, which is crucial for the bioavailability of drugs. It details the mechanisms, theories, and factors affecting the rate of dissolution, highlighting important models like the diffusion layer, Danckwert's model, and the interfacial barrier model. Additionally, it covers the influence of physicochemical properties, formulation aspects, and test parameters on dissolution rates, concluding that in-vitro dissolution studies are vital for ensuring drug bioavailability.