Downloaded 56 times







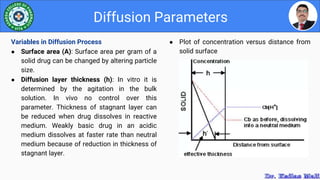



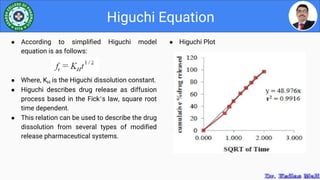






























This document discusses various parameters used to study drug release and dissolution from pharmaceutical dosage forms, including diffusion parameters, dissolution parameters, pharmacokinetic parameters, and models like Higuchi and Peppas plots. It defines key terms like diffusion, flux, Fick's first law, and discusses how factors like agitation, pH, surfactants, viscosity, and temperature can influence dissolution. Key drug release mechanisms and models are also summarized.















































































