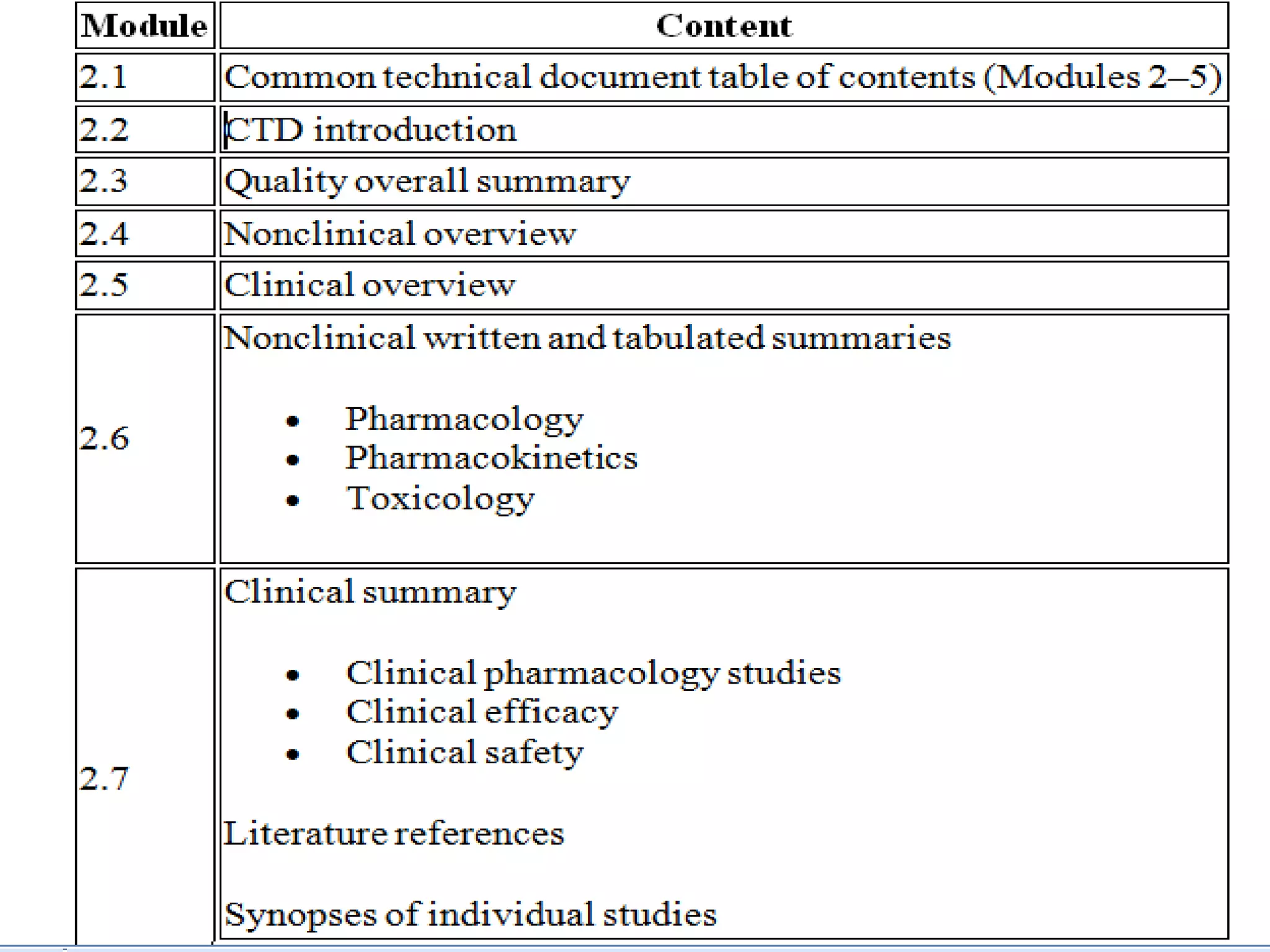
The document discusses the Common Technical Document (CTD) format, which was created by the International Conference on Harmonisation to standardize the submission of documentation for drug approval across regions. It provides a five-module structure for organizing quality, safety, efficacy and other information. While CTD helped streamline submissions, the electronic CTD (eCTD) was later developed to further facilitate electronic transfer and review of documentation between regulators and industry. eCTD utilizes XML formatting and allows lifecycle management of submissions but implementation presents challenges around file formats, regional rules and last minute changes.



































![cmc [ chemistry manufacturing control ]](https://cdn.slidesharecdn.com/ss_thumbnails/presentation2222ra-181120122336-thumbnail.jpg?width=640&height=640&fit=bounds)































![Presentation on the Common Technical Document [CTD] and Electronic Common Te...](https://cdn.slidesharecdn.com/ss_thumbnails/presentationctdandectd-250523171741-67a47446-thumbnail.jpg?width=640&height=640&fit=bounds)











































