Download to read offline







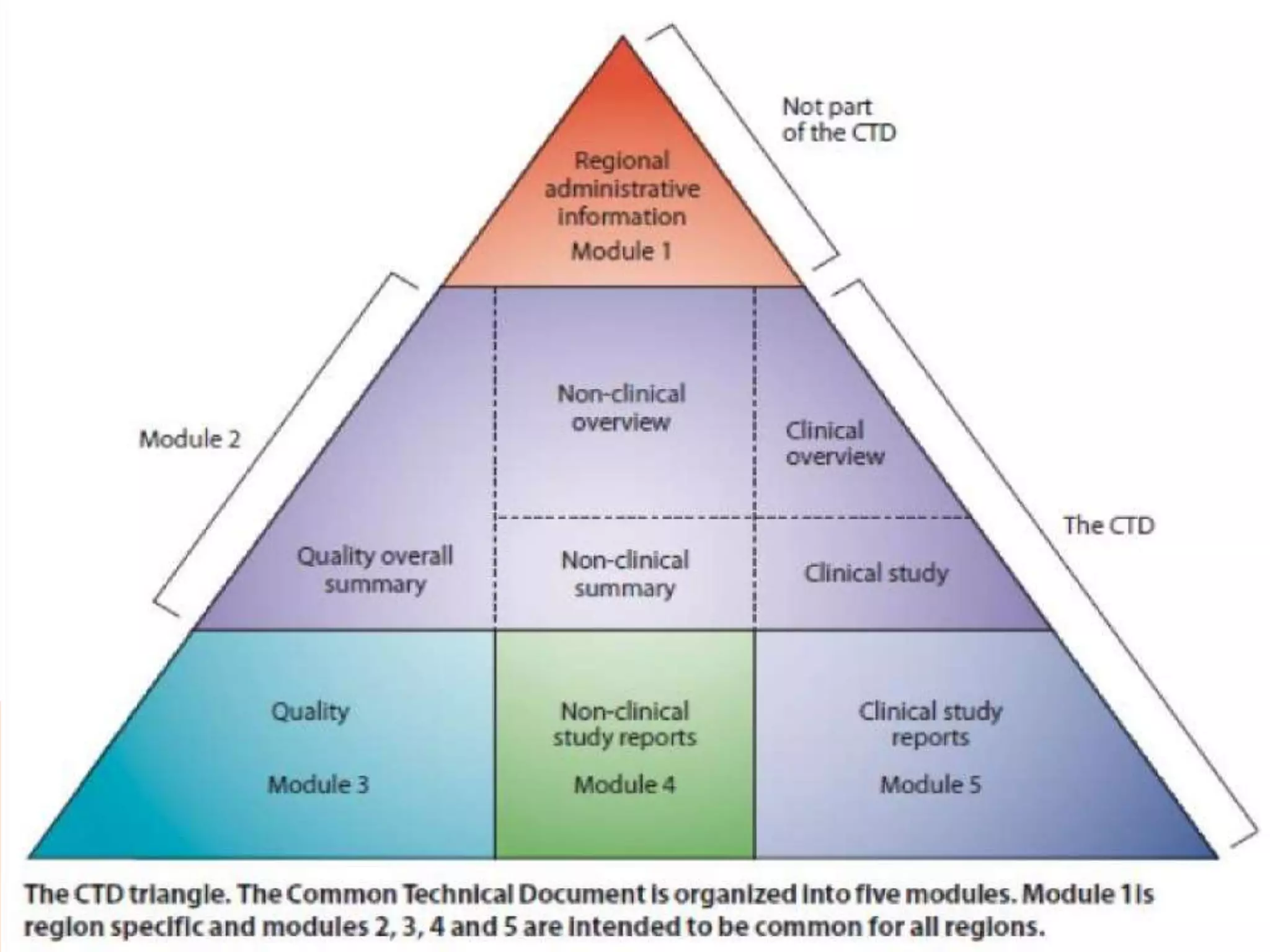

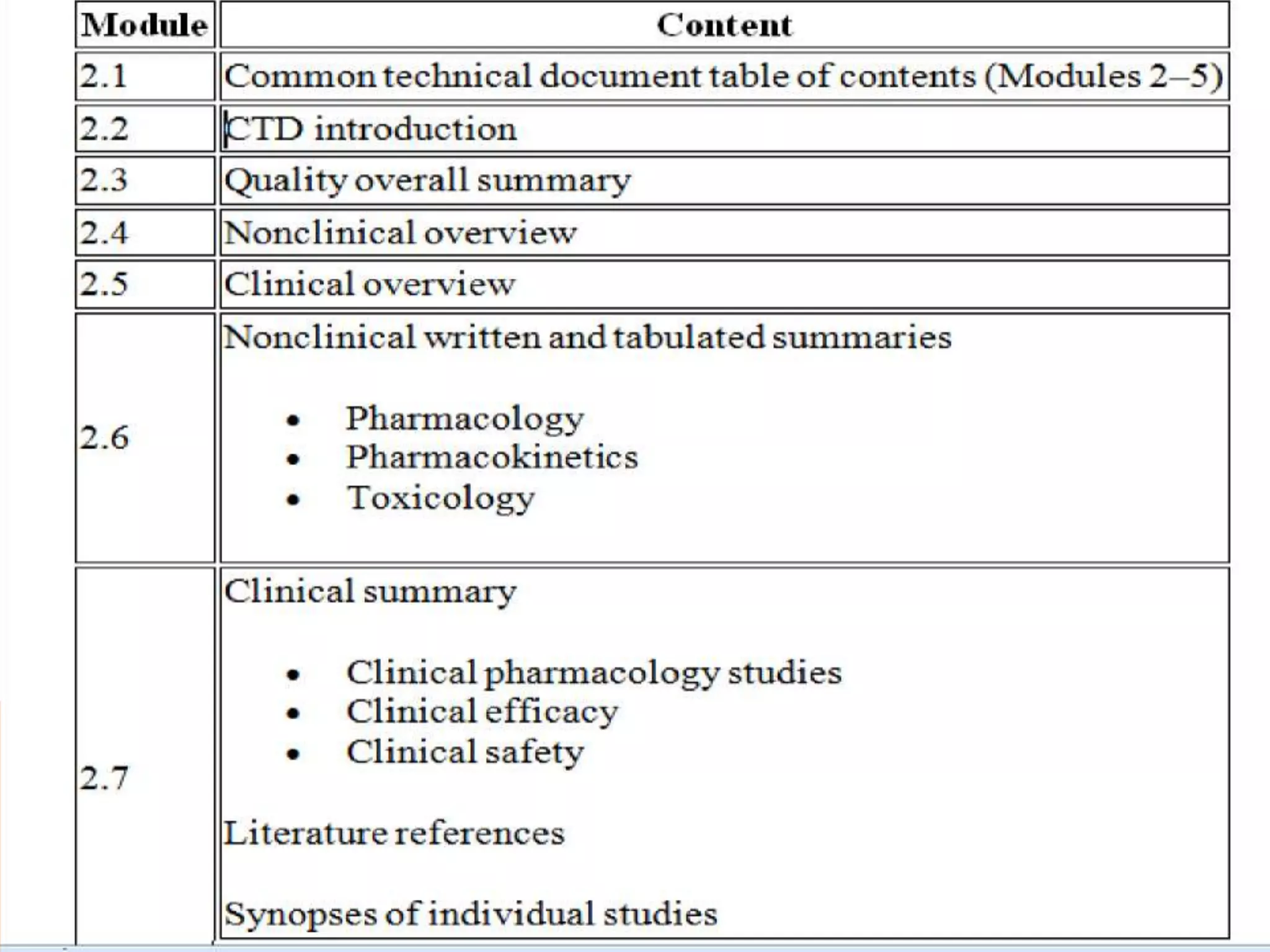

















The document discusses the Common Technical Document (CTD) format for organizing drug regulatory submissions. It notes that CTD was created to standardize submissions across regions and overcome issues like complex approval processes. CTD structures information into five modules covering administrative data, summaries, quality, nonclinical studies, and clinical studies. The document also describes the electronic CTD (eCTD) format and its benefits like improved submission and review processes through use of standardized electronic files and metadata. Implementation challenges include the need for specialized tools and training as well as varying regional requirements.









































![Presentation on the Common Technical Document [CTD] and Electronic Common Te...](https://cdn.slidesharecdn.com/ss_thumbnails/presentationctdandectd-250523171741-67a47446-thumbnail.jpg?width=640&height=640&fit=bounds)


































