Downloaded 22 times
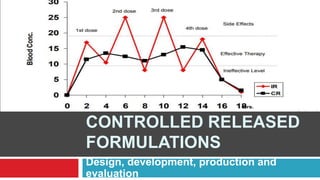

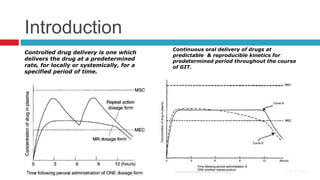
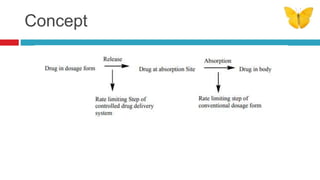
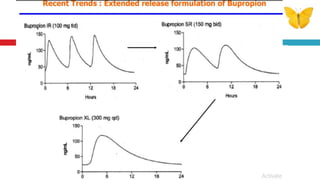












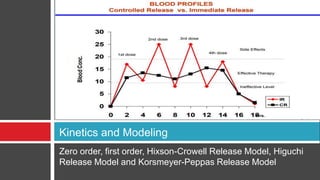


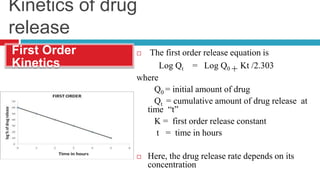

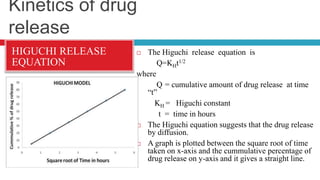
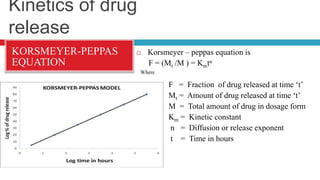

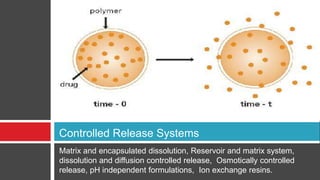


The document outlines the design, development, production, and evaluation of controlled release (CR) formulations, detailing concepts such as drug release kinetics, oral controlled release systems, and evaluation methods. It discusses advantages like improved patient compliance and reduced systemic side effects, while also addressing challenges and disadvantages, including physiological variability and limitations in drug types suitable for CR systems. Key factors influencing CR product performance include physicochemical properties, pharmacokinetics, and biological factors.




















































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)
