1
INSTITUTE OF PHARMACEUTICALSCIENCES, KUK
(SESSION 2025-2027)
Subject: Drug Delivery System
(MPH 102T)
TOPIC- GENERAL INTRODUCTION OF SUSTAINED RELEASE AND CONTROLLED RELEASE
Submitted to:-Mrs. Geeta Jangra
Presented by:- Taniya
M.Pharm 1st
Semester
2.
2
CONTENT
1) Introduction andbasic concepts of SR and CR
2) Advantages
3) Disadvantages
4) Difference between sustained and controlled release
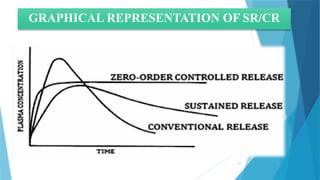
5) Graphical Representation of CR and SR
6) Factors affecting SR/CR
7) Physiochemical and biological approaches for SR/CR formulations
8) Mechanism of Drug Delivery from SR/CR Formulations
3.
3
INTRODUCTION TO SR/CRFORMULATIONS
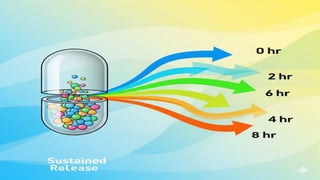
SUSTAINED RELEASE DRUG FORMULATIONS
Sustained release dosage forms are the formulations that release drug over and extended
period, maintaining the therapeutic drug levels in the body. There is a slow release over a
long period maintaining its effect for a long time.
It helps to reduce dosing frequency, improving patient compliance and is potential for
improved efficacy.
It follows first order where rate of release decreases over time as the drug is depleted.
For example- Aspirin, Dextrin SR tablets, implants etc.
5
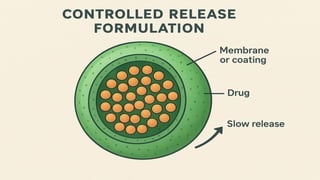
CONTROLLED RELEASE DRUGFORMULATIONS
Controlled release dosage forms are the formulations that deliver drug at a specific
release rate within a predetermined time period thus maintaining consistent drug
concentration in the body.
It helps to improve therapeutic efficacy, reduce side effects and target delivery to specific
sites.
It follow zero order kinetics.
For example- insulin pumps, transdermal patches etc.
7
ADVANTAGES
Reduced dosingfrequency
Dose reduction
Improved patient compliance
A constant level of drug concentration in blood plasma
Reduced toxicity due to overdose
Reduces the fluctuation of peak-valley concentration
Night time dosing can be avoided
Economic
The total amount of drug administered can be reduced, thus:
1. Maximizing availability with minimum dose
2. Minimize or eliminate local side effects
3. Minimize or eliminate systemic side effects
4. Minimize drug accumulation with chronic dosing.
8.
8
DISADVANTAGES
Probability of dosedumping
Reduced potential for dose adjustment
Cost of single unit higher than conventional dosage forms
Increase potential for first-pass metabolism
The requirement for additional patient education for proper medication
Decreased systemic availability in comparison to immediate release
conventional dosage forms
Poor in vitro and in vivo correlations.
9.
9
DIFFERENCE BETWEEN SR/CR
SUSTAINEDRELEASE
Provide medication over extended
period of time.
Follows 1st
order release kinetics.
Do not promote localization of drugs
at active sites.
Drug release at prolonged rate
dependent on external environment.
CONTROLLED RELEASE
Maintain constant drug levels in the
blood/tissue
Follow zero order release kinetics.
Promote localization of the drug at
active sites.
Drug release at predetermined rate
independent of the external
environment.
11
Pharmacokinetic & PharmacodynamicProperties of SR/CR
Formulations
Pharmacokinetic properties:-
Absorption
• Slower and
prolonged
compared to
immediate
release (IR).
• Designed to
maintain
therapeutic
drug levels
for extended
periods.
• Reduced
peak–trough
fluctuations
Distribution
• Generally not
altered by
SR/CR
formulation
itself.
• Steady plasma
concentration
can improve
predictability
of
distribution.
Metabolism
• Reduced
chances of
first-pass
metabolism
overload (due
to slower
release).
• Some drugs
may show
altered
bioavailability
depending on
release
mechanism.
Excretion
• More constant
drug plasma
levels →
predictable
elimination.
• Dosing
frequency
reduced,
minimizing
accumulation
risk.
Half-life
• Apparent
half-life may
appear longer
because of
prolonged
release, even
though the
drug’s true
biological
half-life
remains the
same.
Bioavailability
• Can be
increased or
decreased
depending on
drug
properties and
formulation.
• More
consistent
systemic
availability
compared to
immediate
release
12.
12
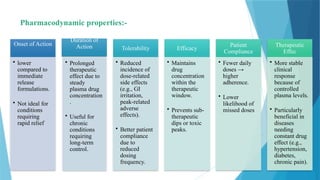
Pharmacodynamic properties:-
Onset ofAction
• lower
compared to
immediate
release
formulations.
• Not ideal for
conditions
requiring
rapid relief
Duration of
Action
• Prolonged
therapeutic
effect due to
steady
plasma drug
concentration
.
• Useful for
chronic
conditions
requiring
long-term
control.
Tolerability
• Reduced
incidence of
dose-related
side effects
(e.g., GI
irritation,
peak-related
adverse
effects).
• Better patient
compliance
due to
reduced
dosing
frequency.
Efficacy
• Maintains
drug
concentration
within the
therapeutic
window.
• Prevents sub-
therapeutic
dips or toxic
peaks.
Patient
Compliance
• Fewer daily
doses →
higher
adherence.
• Lower
likelihood of
missed doses
Therapeutic
Effec
• More stable
clinical
response
because of
controlled
plasma levels.
• Particularly
beneficial in
diseases
needing
constant drug
effect (e.g.,
hypertension,
diabetes,
chronic pain).
13.
13
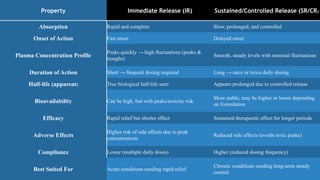
Property Immediate Release(IR) Sustained/Controlled Release (SR/CR)
Absorption Rapid and complete Slow, prolonged, and controlled
Onset of Action Fast onset Delayed onset
Plasma Concentration Profile
Peaks quickly → high fluctuations (peaks &
troughs)
Smooth, steady levels with minimal fluctuations
Duration of Action Short → frequent dosing required Long → once or twice daily dosing
Half-life (apparent) True biological half-life seen Appears prolonged due to controlled release
Bioavailability Can be high, but with peaks/toxicity risk
More stable, may be higher or lower depending
on formulation
Efficacy Rapid relief but shorter effect Sustained therapeutic effect for longer periods
Adverse Effects
Higher risk of side effects due to peak
concentrations
Reduced side effects (avoids toxic peaks)
Compliance Lower (multiple daily doses) Higher (reduced dosing frequency)
Best Suited For Acute conditions needing rapid relief
Chronic conditions needing long-term steady
control
14.
14
FACTORS AFFECTING THESR/CR FORMULATION
PHYSICOCHEMICAL FACTORS
Aqueous solubility
Partition coefficient (P [O/W])
Drug pKa and ionization at physiological pH
Drug stability
Molecular weight and diffusivity
Drug–Excipient Interaction
Dose size.
BIOLOGICAL FACTORS
Absorption
Distribution
Metabolism
Biological half-life/duration of action
Margin of safety/therapeutic index
Side effect
Disease state.
15.
15
Physiochemical factors
• Effect:Drugs with very low solubility dissolve slowly → incomplete
release. Highly soluble drugs dissolve too fast → burst release.
• Suitable: Moderately soluble drugs (ideal balance).
• Unsuitable: Very low (<0.1 mg/ml) or very high (>10 mg/ml)
solubility drugs.
Aqueous
Solubility
• Effect: Determines drug permeability across GI membrane.
• Suitable: Drugs with moderate lipophilicity (log P ~1–3).
• Unsuitable: Very hydrophilic (poor permeability) or very lipophilic
(poor solubility).
Partition
Coefficient
• Effect: Weak acids/bases show pH-dependent solubility → variable
release in different GI regions.
• Suitable: Drugs with pH-independent solubility near physiological
pH.
• Unsuitable: Drugs with extreme pH-dependent solubility (risk of
erratic release).
Ionization &
pKa
16.
16
• Effect: Largermolecules diffuse slowly through polymer/matrix →
incomplete release.
• Suitable: Small to moderately sized molecules.
• Unsuitable: High molecular weight drugs (proteins, peptides).
Molecular Size
& Diffusivity
• Effect: Large doses require bulky SR dosage forms → difficult for
patient compliance.
• Suitable: Potent drugs requiring ≤500 mg/day.
• Unsuitable: High-dose drugs (>1 g/day).
Dose Size
• Effect: Chemical or physical interactions with polymers can alter
release profile.
• Suitable: Drugs chemically compatible with common SR polymers
(HPMC, ethylcellulose, etc.).
• Unsuitable: Reactive or unstable drugs with excipients
Drug–
Excipient
Interaction
• Effect: Drugs unstable in GI fluids (acid/base hydrolysis, enzymatic
degradation) may degrade before absorption.
• Suitable: Stable drugs throughout GI tract.
• Unsuitable: Unstable drugs (e.g., penicillin G, peptides)
Stability
17.
17
Biological factors
Absorption
• Effect:The absorption rate and site play a major role. If a drug is absorbed only from a specific part
of the GI tract (e.g., upper intestine), SR/CR may fail because the dosage form stays longer in the
gut.
• Suitable drugs: Drugs with good absorption throughout the GIT.
• Not suitable: Drugs with absorption "window" (like levodopa, riboflavin).
Distribution
• Effect: Wide tissue distribution lowers plasma concentration quickly, making it harder to
maintain steady therapeutic levels with SR/CR.
• Suitable drugs: Drugs with limited distribution (small Vd).
• Not suitable: Drugs with very high tissue binding (large Vd).
Metabolism
• Effect: Drugs with extensive first-pass metabolism are problematic, as a slow release may
increase metabolism and reduce bioavailability.
• Suitable drugs: Moderately metabolized drugs.
• Not suitable: Drugs with very high first-pass effect (e.g., propranolol, nitroglycerin).
18.
18
Biological Half-life /Duration of Action
• Effect: Half-life is critical for SR/CR.
• Suitable drugs: Half-life of 2–6 hours (good balance for sustained release).
• Not suitable:
• Very short half-life (<1 hr) → drug elimination too rapid.
• Very long half-life (>12 hrs) → sustained release unnecessary.
Margin of Safety / Therapeutic Index
• Effect: Narrow therapeutic index drugs are risky, because small changes in release rate → toxicity or sub-
therapeutic effect.
• Suitable drugs: Wide therapeutic index (safe margin).
• Not suitable: Narrow TI drugs (e.g., digoxin, lithium, theophylline – though some still used with
caution).
Side Effects
• Effect: Drugs causing GI irritation (e.g., aspirin, NSAIDs) are unsuitable, as SR prolongs contact with
mucosa.
• Suitable drugs: Drugs with minimal local irritation.
• Not suitable: Drugs causing severe GI irritation/toxicity.
Disease State
Effect: Pathological conditions can alter absorption, metabolism, or clearance → unpredictable drug release.
Examples:
Diarrhea, Crohn’s disease → decreased absorption.
Hepatic disease → altered metabolism.
Renal impairment → slower clearance, risk of toxicity.
19.
19
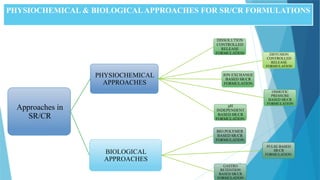
PHYSIOCHEMICAL & BIOLOGICALAPPROACHESFOR SR/CR FORMULATIONS
Approaches in
SR/CR
PHYSIOCHEMICAL
APPROACHES
DISSOLUTION
CONTROLLED
RELEASE
FORMULATION DIFFUSION
CONTROLLED
RELEASE
FORMULATION
ION EXCHANGE
BASED SR/CR
FORMULATION
OSMOTIC
PRESSURE
BASED SR/CR
FORMULATION
pH
INDEPENDENT
BASED SR/CR
FORMULATION
BIOLOGICAL
APPROACHES
BIO POLYMER
BASED SR/CR
FORMULATION
PULSE BASED
SR/CR
FORMULATION
GASTRO
RETENTION
BASED SR/CR
FORMULATION
20.
20
Physiochemical approaches
1.Matrix Systems
Polymer-basedmatrices: These involve embedding the drug in a
polymer matrix that controls drug release through diffusion and/or
polymer degradation.
Hydrophilic matrices: Water-soluble polymers like
hydroxypropylmethylcellulose (HPMC) or
polyvinylpyrrolidone (PVP) absorb water and swell, creating a
gel layer that controls drug release.
Hydrophobic matrices: These are made from lipophilic
polymers, such as ethyl cellulose, which release the drug
primarily through diffusion.
2.Reservoir Systems
Membrane-controlled: In this type of system, the drug is
encapsulated in a core surrounded by a semi-permeable
membrane. Drug release occurs via diffusion through the
membrane. Examples include osmotic pumps and lipid-based
delivery systems.
Osmotic pump systems: These utilize an osmotic pressure
difference to drive drug release. Water enters the device, dissolves
the drug, and the solution is released through a small orifice.
21.
21
3.Ion Exchange Systems
These systems use ion-exchange resins to bind the drug and release it in response to changes in the pH or ionic
strength of the surrounding environment (e.g., the gastrointestinal tract).
Example: Sodium-calcium resins can be used for drugs that are weak acids or bases.
4.Liposomes and Nanoparticles
Liposomes: These lipid-based vesicles encapsulate the drug, allowing for controlled release. They are particularly
useful for both hydrophilic and lipophilic drugs.
Nanoparticles: Drug-loaded nanoparticles (e.g., polymeric nanoparticles, nanostructured lipid carriers (NLCs))
can offer controlled drug release through size, surface charge, and polymer type manipulation.
5.Hydrogels
Hydrogels are three-dimensional networks of hydrophilic polymers that absorb water and swell. The drug release can
be controlled by factors such as pH, temperature, or ionic strength.
Common polymers used: Polyethylene glycol (PEG), Polyacrylamide, and Polyvinyl alcohol (PVA).
22.
Biological approaches
1.Enzyme-based DeliverySystems
Enzyme-sensitive polymers can be used to design systems that release drugs only when exposed to
specific enzymes (e.g., lipase-sensitive systems or protease-sensitive polymers).
This is particularly useful for targeting specific sites within the body (e.g., the gastrointestinal tract) where
the enzymes are prevalent.
2.Biodegradable Polymers
Polymers that degrade over time, such as polylactic acid (PLA), poly(lactic-co-glycolic acid) (PLGA),
and polycaprolactone (PCL), can be used for SR/CR formulations. The degradation of the polymer
matrix leads to the gradual release of the drug.
PLGA-based formulations are commonly used for injectable controlled-release formulations and
implants.
3.Protein or Peptide Drug Delivery
Biodegradable microparticles or nanoparticles can encapsulate protein or peptide drugs, protecting
them from enzymatic degradation and allowing for slow release over time. This is particularly important
for biologic therapies where protein stability is a concern.
22
23.
23
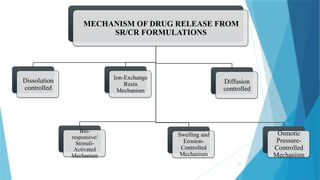
MECHANISM OF DRUGRELEASE FROM
SR/CR FORMULATIONS
Dissolution
controlled
Bio-
responsive/
Stimuli-
Activated
Mechanism
Ion-Exchange
Resin
Mechanism
Swelling and
Erosion-
Controlled
Mechanism
Diffusion
controlled
Osmotic
Pressure-
Controlled
Mechanism
24.
24
Drug diffuses througha polymer matrix or a membrane at a controlled rate.
Types:
Reservoir system → Drug is enclosed in a polymer coat; release occurs by diffusion through the membrane.
Matrix system → Drug is uniformly dispersed in a polymer/lipid matrix; release occurs as drug diffuses through the
pores.
Example: Ethylcellulose-coated tablets, polymeric films.
2.Dissolution-Controlled Mechanism
Drug release depends on the dissolution rate of the polymer or the drug itself.
Types:
Dissolution of drug → Poorly soluble drug dissolves slowly.
Dissolution of coating → Slowly dissolving polymer coat controls release.
Example: Coated tablets with cellulose acetate phthalate.
1.Diffusion-Controlled Mechanism
25.
25
3. Ion-Exchange ResinMechanism
In this drug is complexed with an insoluble ion-exchange resin. The drug is bound to the resin and released by exchanging
with the ion present in the GI fluid.
This mechanism is influenced by the concentration of the ions and the pH of the environment.
This system is designed to provide the controlled release of the ionizable drug.
Resin+ - drug− + X− →→ Resin+ - X− + drug-
Resin− - drug+ + Y+ →→ Resin− - Y+ + drug+
Where, X− and Y+ are ions the GI tract.
The rate of drug diffusing out of the resin is controlled by the area of diffusion, diffusion path length, and rigidity of the resin,
which is the function of the amount of cross-linking agent used to prepare the resin. For the better release in this system is to
coat the ion-exchange resin with hydrophobic rate-limiting polymer.
Example: Polystyrene sulfonate resins with drugs like dextromethorphan.
4.Swelling and Erosion-Controlled Mechanism
Hydrophilic polymers (HPMC, Carbopol) swell in contact with GI fluids.
Gel layer forms → controls diffusion of drug.
Later, polymer erodes → releasing more drug.
Example: Hydrophilic matrix tablets.
26.
26
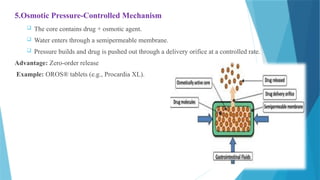
5.Osmotic Pressure-Controlled Mechanism
The core contains drug + osmotic agent.
Water enters through a semipermeable membrane.
Pressure builds and drug is pushed out through a delivery orifice at a controlled rate.
Advantage: Zero-order release
Example: OROS® tablets (e.g., Procardia XL).









![14
FACTORS AFFECTING THE SR/CR FORMULATION
PHYSICOCHEMICAL FACTORS
Aqueous solubility
Partition coefficient (P [O/W])
Drug pKa and ionization at physiological pH
Drug stability
Molecular weight and diffusivity
Drug–Excipient Interaction
Dose size.
BIOLOGICAL FACTORS
Absorption
Distribution
Metabolism
Biological half-life/duration of action
Margin of safety/therapeutic index
Side effect
Disease state.](https://image.slidesharecdn.com/vishalkumar-251118151514-07b29ad5/85/Controlled-drug-delivery-system-and-pptx-14-320.jpg)


































![Sustained release dosage form [srdf]](https://cdn.slidesharecdn.com/ss_thumbnails/sustainedreleasedosageformsrdf-160516130442-thumbnail.jpg?width=640&height=640&fit=bounds)






















