Downloaded 1,845 times



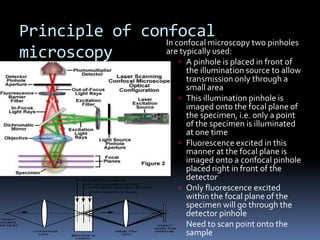
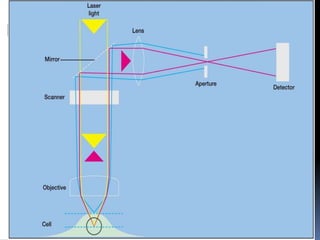

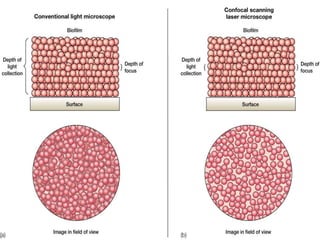



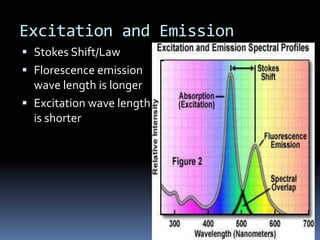
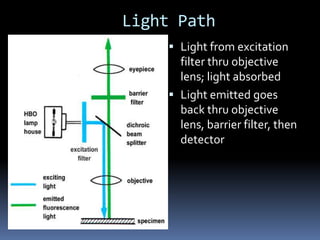


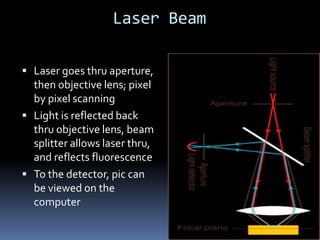









This document provides information about confocal microscopy. It discusses: - How confocal microscopy works by excluding light from out-of-focus planes to generate high-contrast images with better resolution than conventional microscopes. - The history of confocal microscopy, which was pioneered by Marvin Minsky in 1955 using pinholes and point-by-point illumination. - Key aspects of confocal microscopy like using fluorophores, laser excitation, and building 3D images by combining thin optical sections.



































































