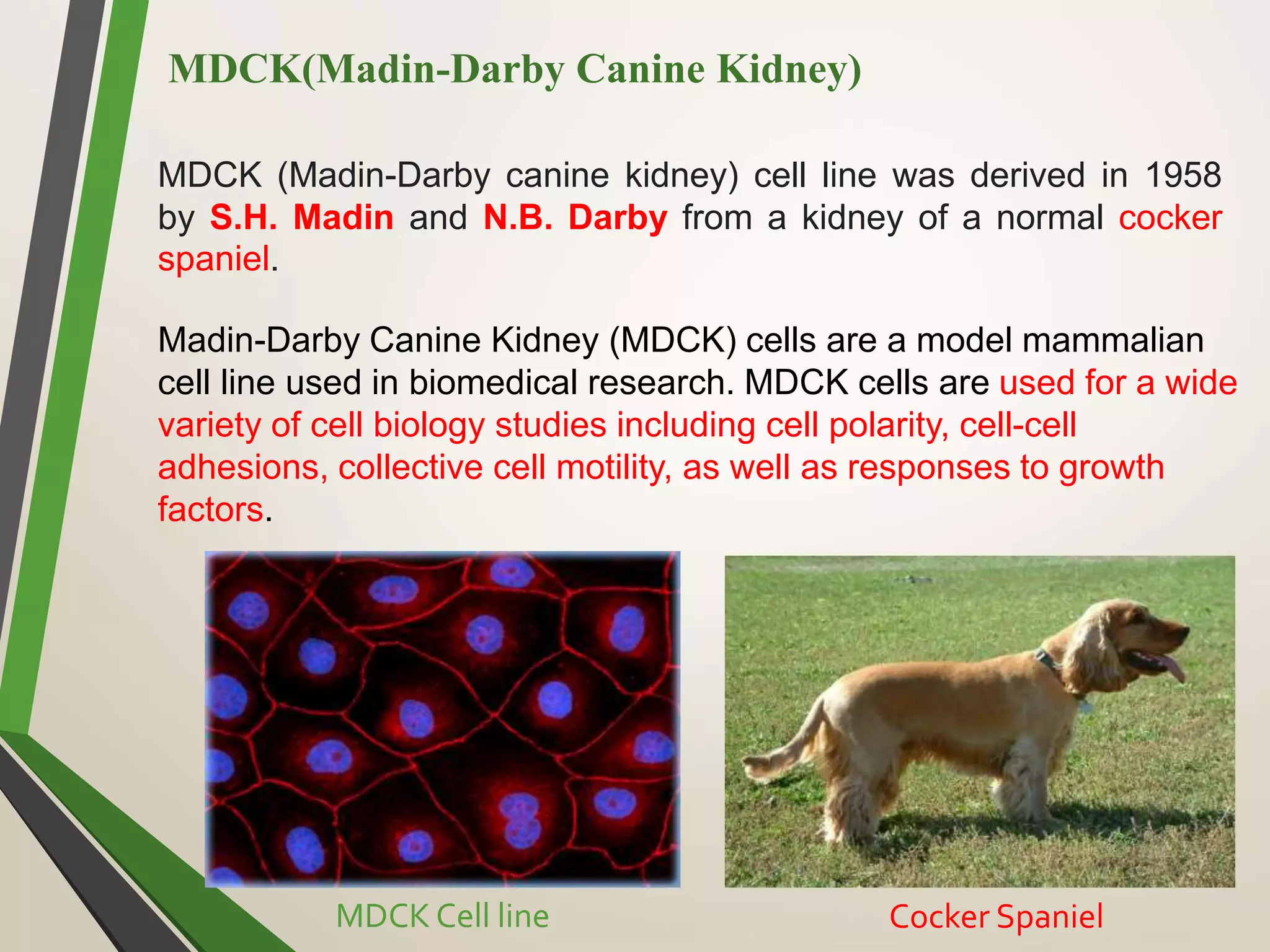
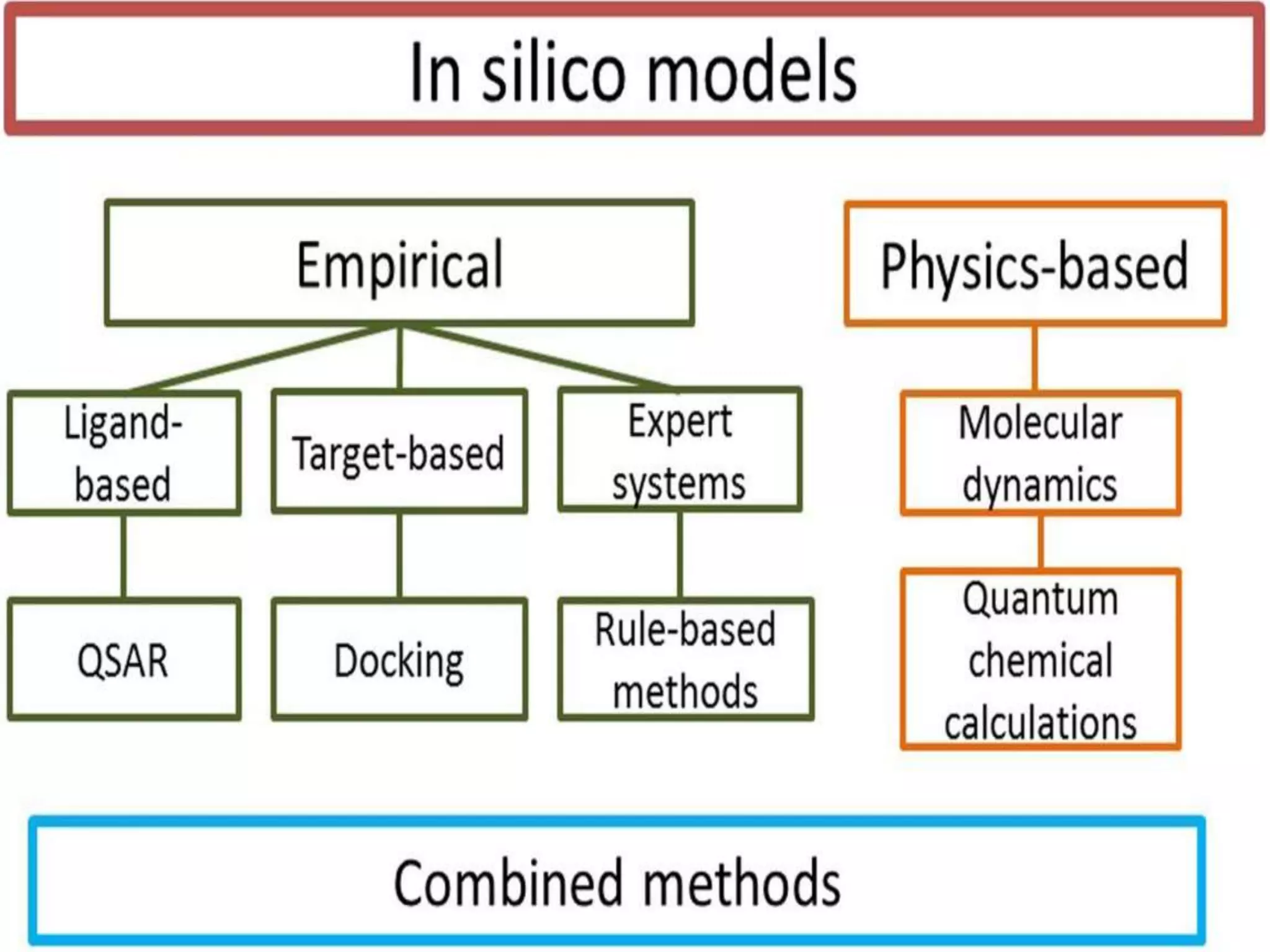
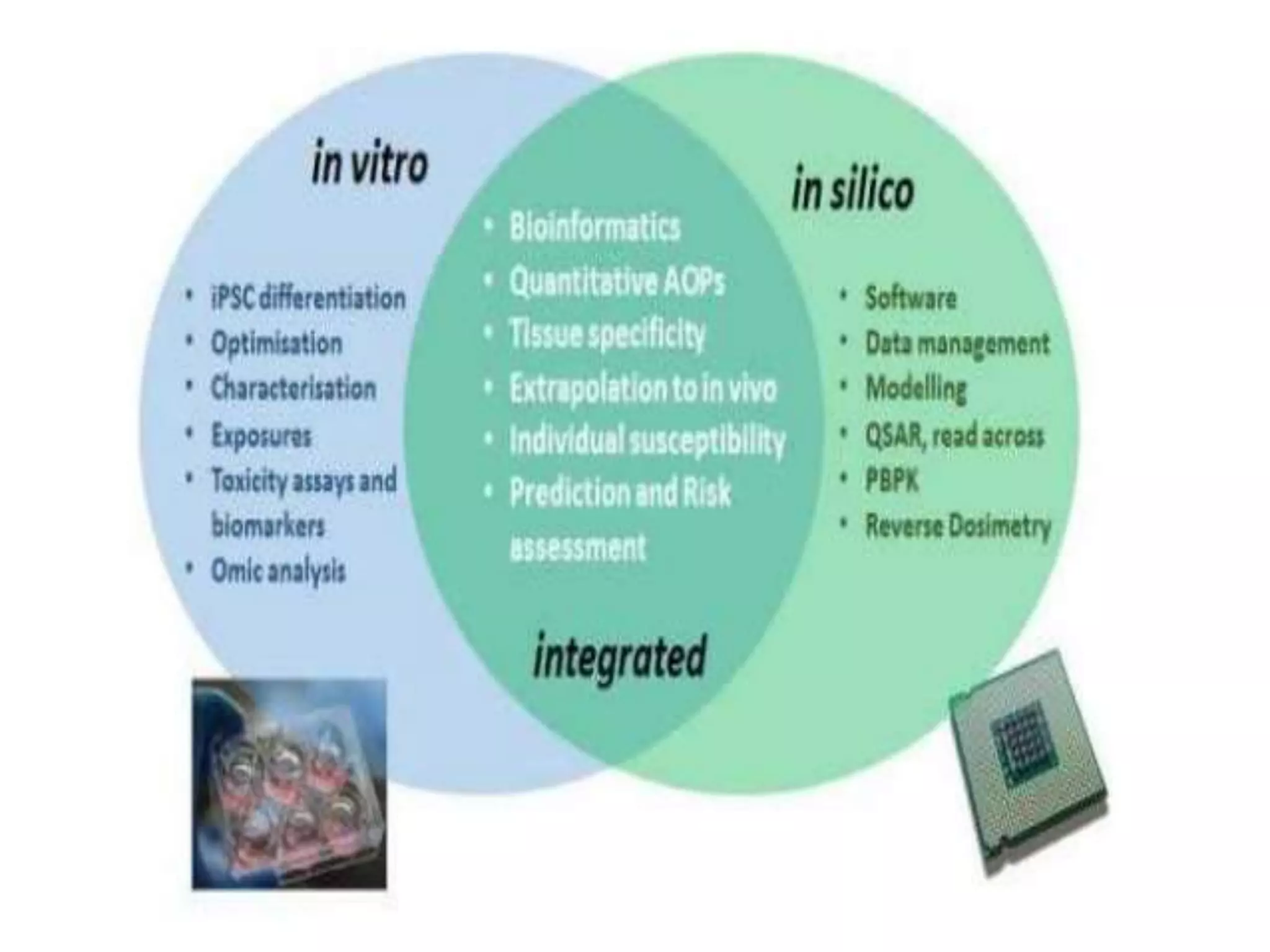
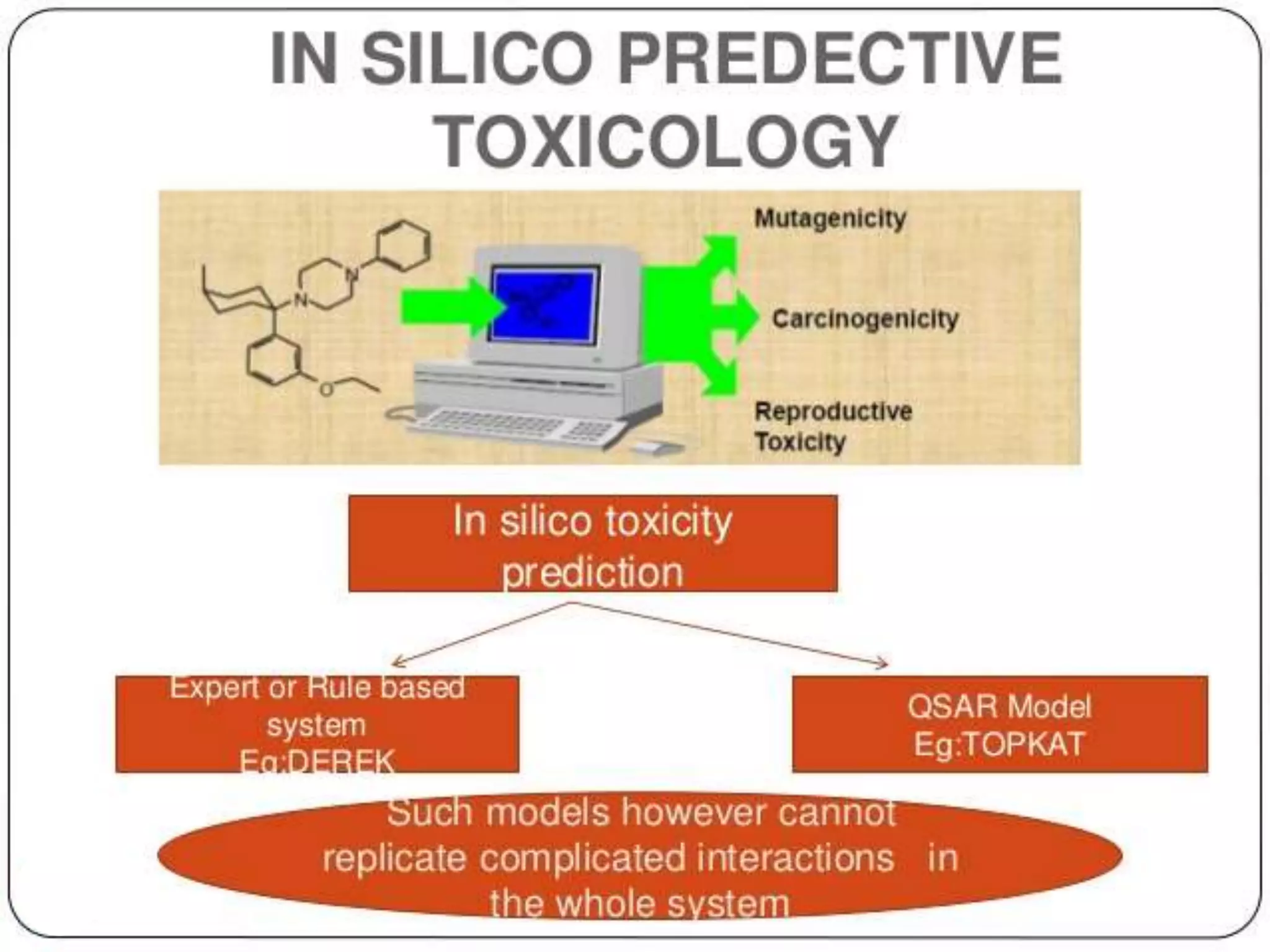
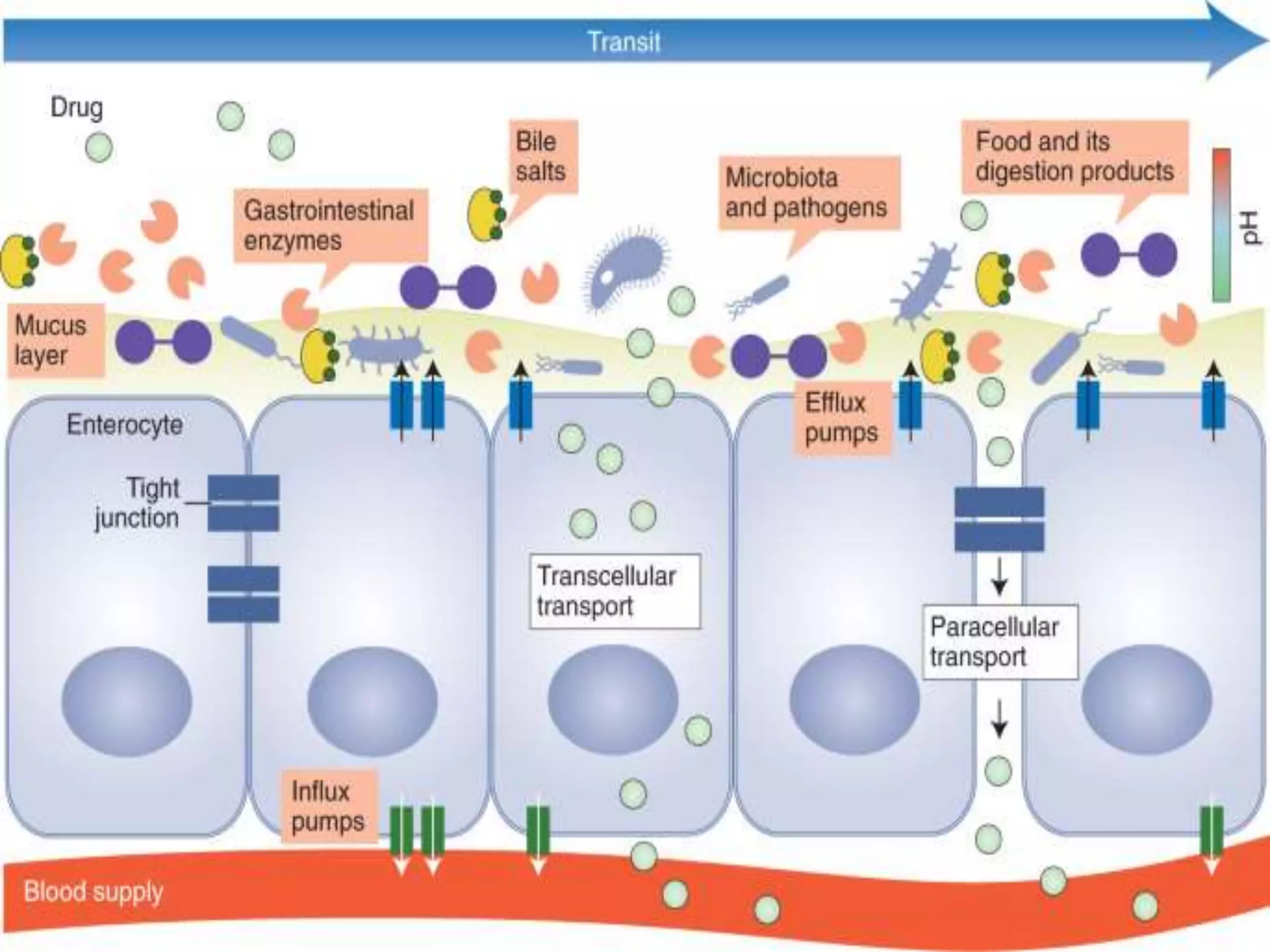
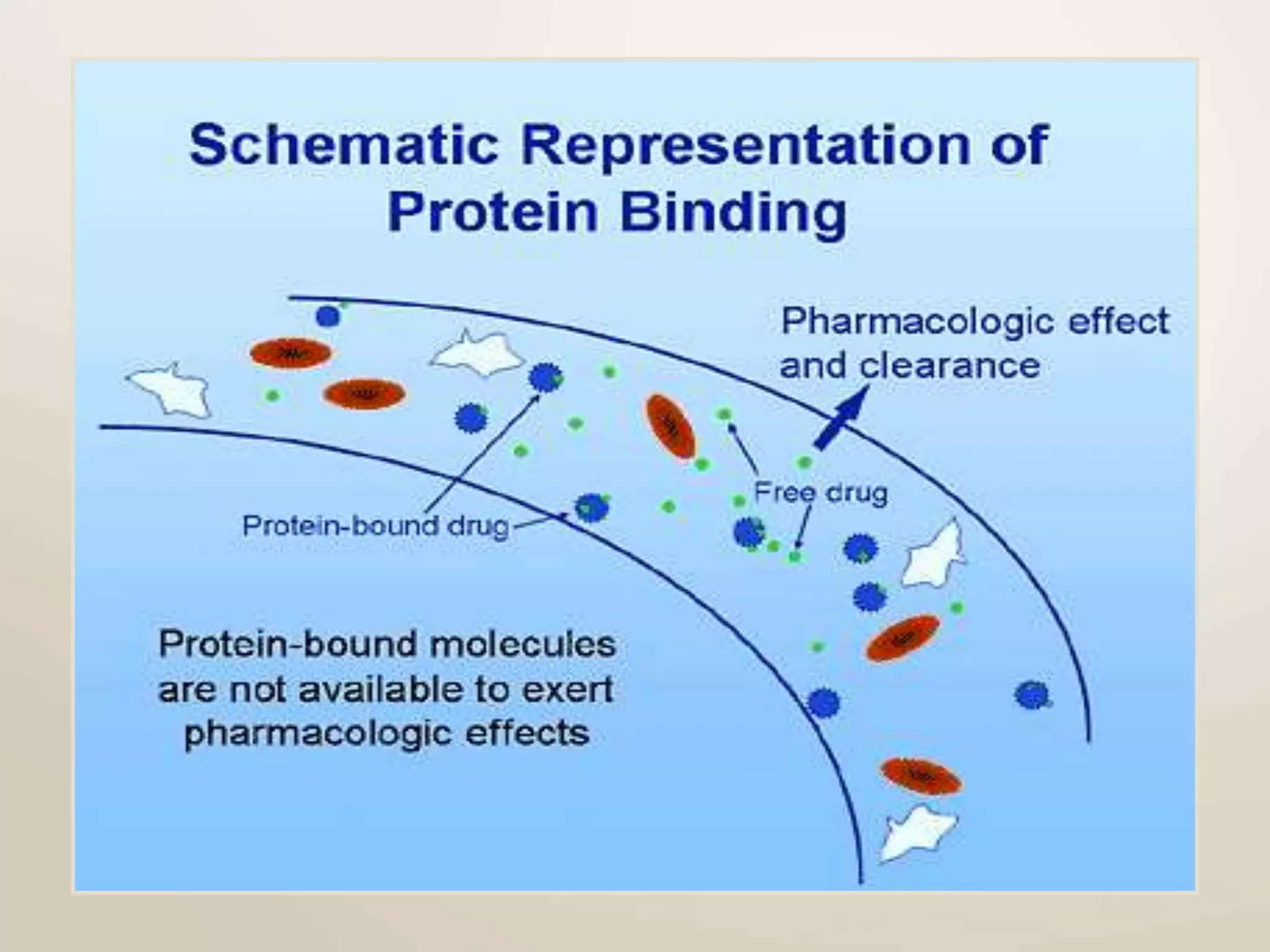
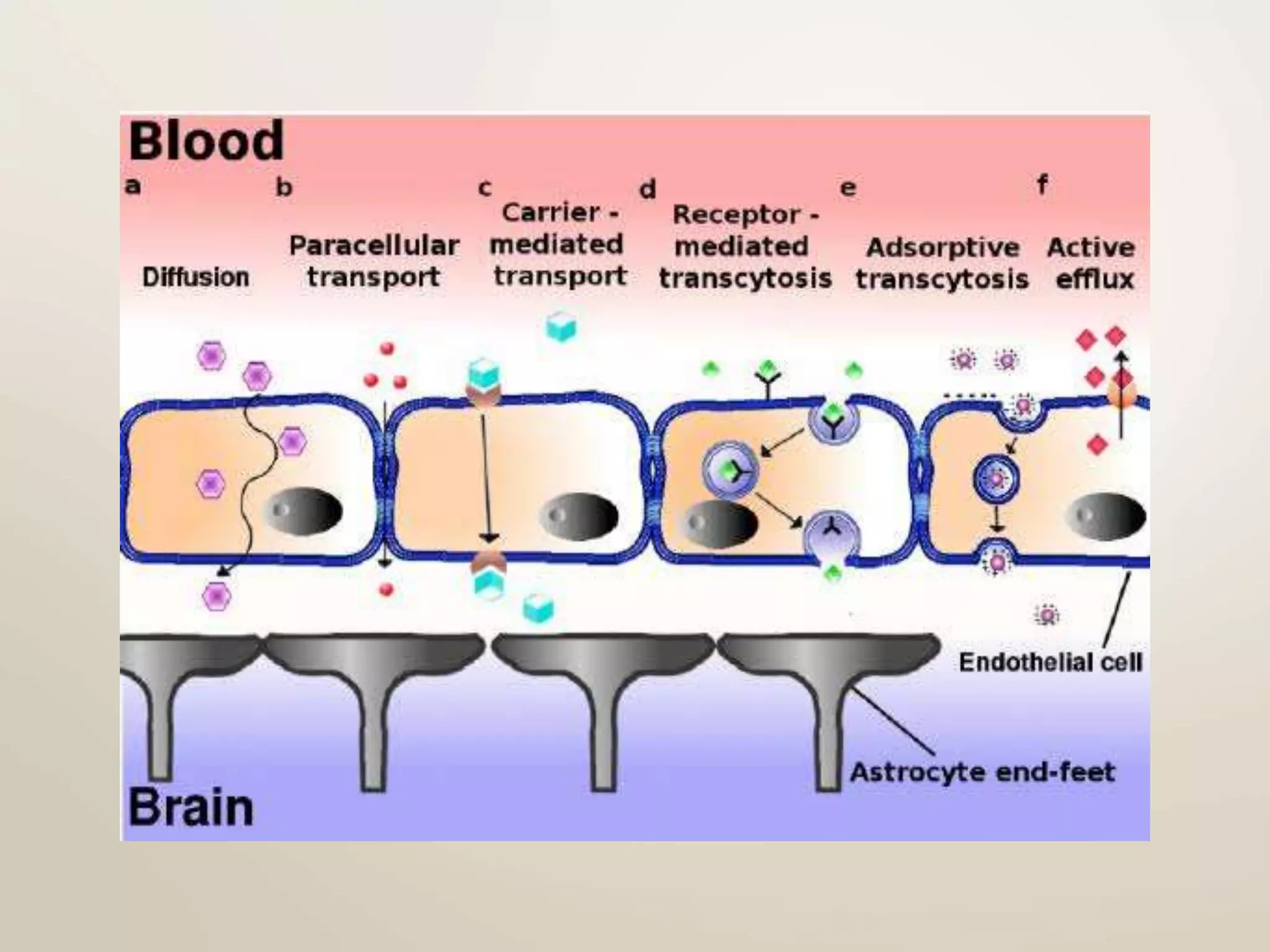
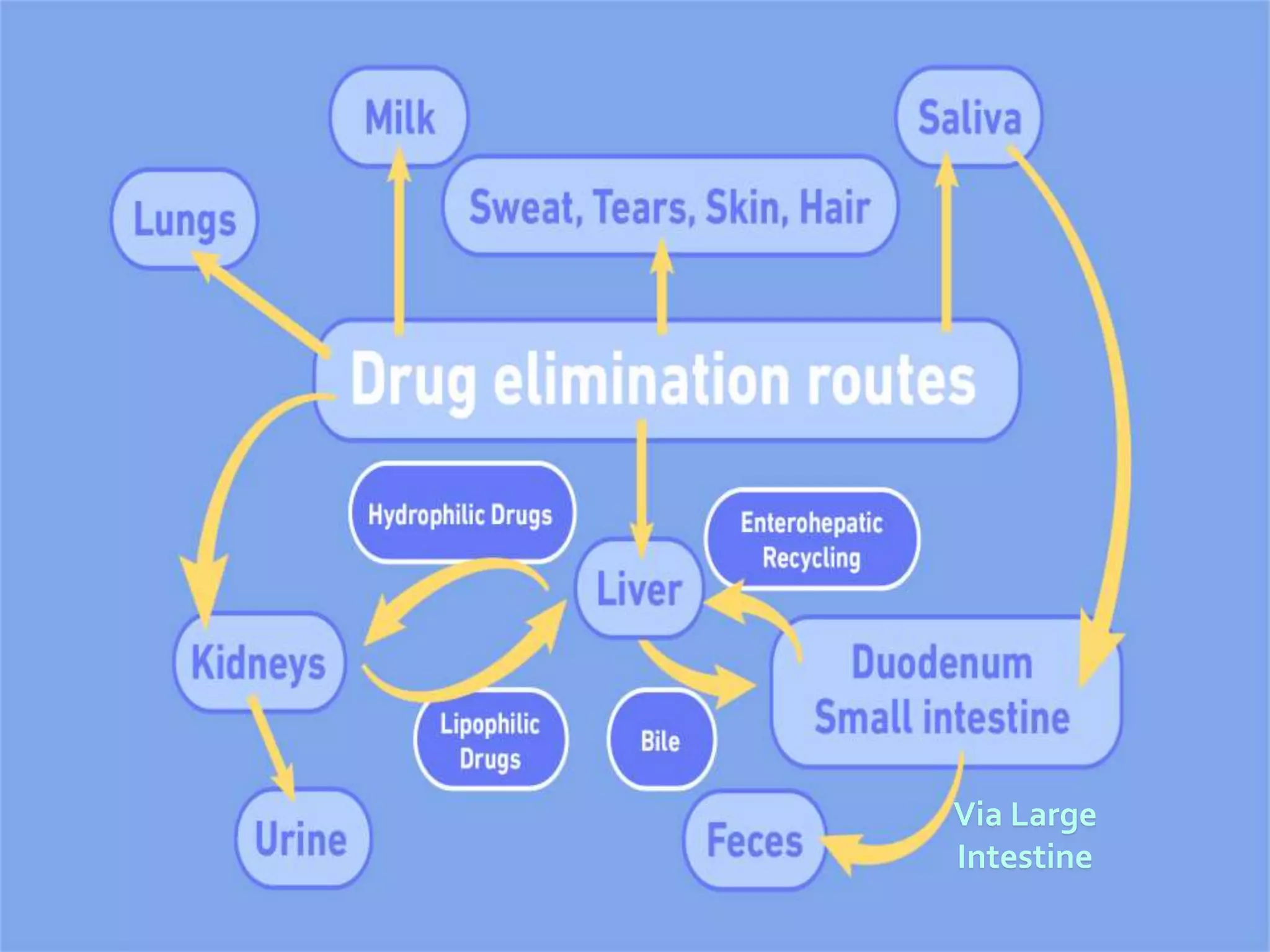
Computational modeling is used to predict drug absorption, distribution, metabolism, excretion and toxicity (ADMET) properties based on a drug's structure. Caco-2 and MDCK cell lines are used to simulate intestinal permeability for predicting oral absorption. Quantitative structure-activity relationship (QSAR) models correlate molecular descriptors with ADMET properties. Absorption models consider solubility, permeability through cell lines like Caco-2, and transporters. Distribution is reflected by volume of distribution, plasma protein binding and blood-brain barrier permeability. Excretion involves hepatic and renal clearance but current models cannot predict clearance directly from structure.





























































![Computational modeling in_drug_disposition[1]](https://cdn.slidesharecdn.com/ss_thumbnails/computationalmodelingindrugdisposition1-200604161524-thumbnail.jpg?width=640&height=640&fit=bounds)































