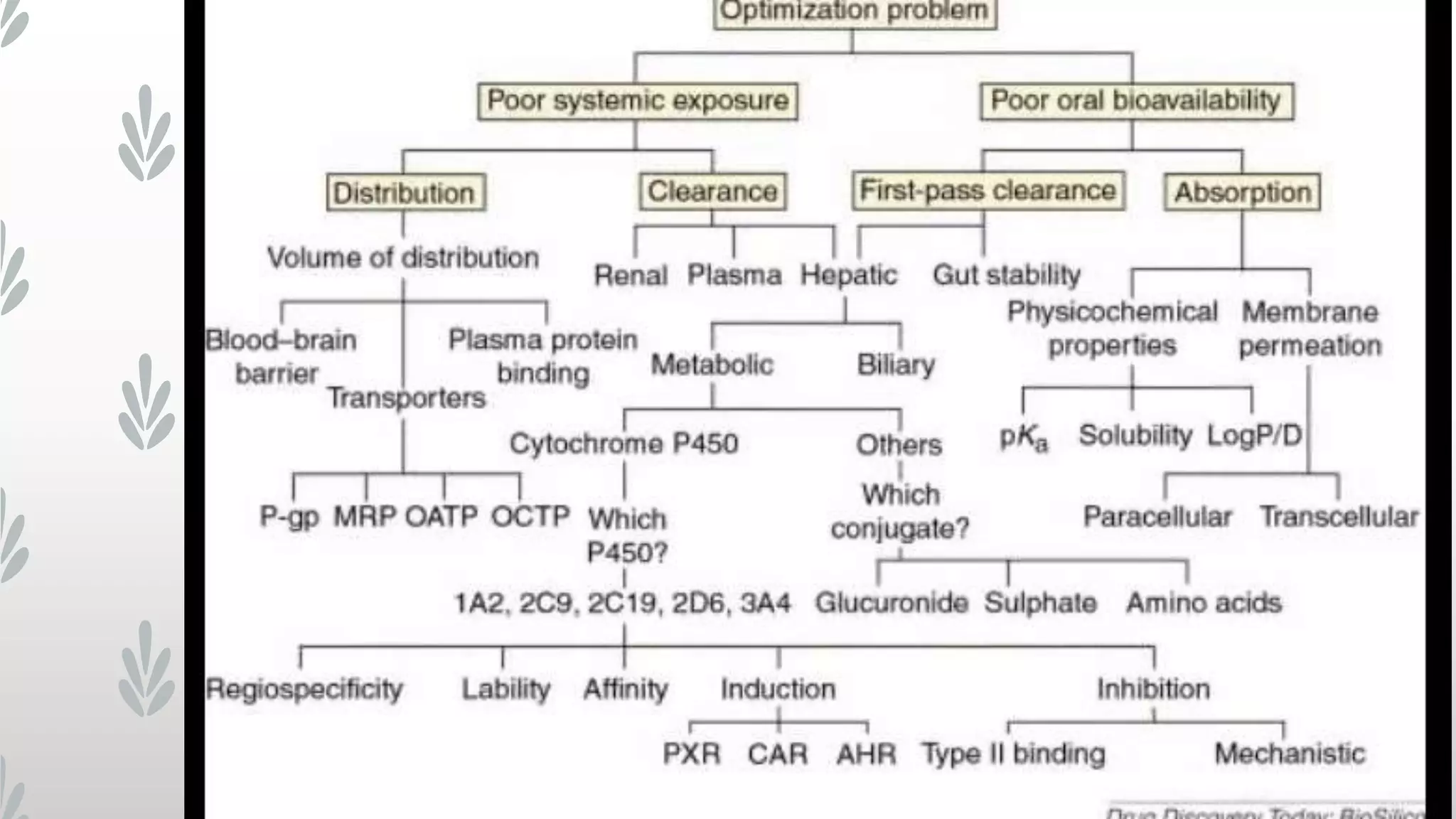
This document discusses computational modeling techniques for predicting drug distribution properties. It covers two main modeling approaches: quantitative approaches like pharmacophore modeling and docking to study drug-target interactions, and qualitative approaches like QSAR and QSPR studies that use multivariate analysis to correlate molecular descriptors with properties. Key aspects of drug distribution addressed include volume of distribution, plasma protein binding, and blood-brain barrier permeability. The challenges of developing accurate predictive models for these properties are also noted.
























































![Computational modeling in_drug_disposition[1]](https://cdn.slidesharecdn.com/ss_thumbnails/computationalmodelingindrugdisposition1-200604161524-thumbnail.jpg?width=640&height=640&fit=bounds)



























