Downloaded 94 times























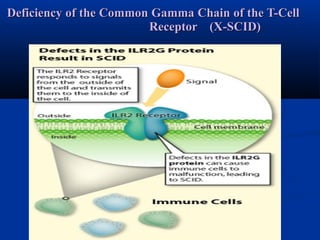






















































The document discusses severe combined immunodeficiency (SCID), a group of fatal disorders characterized by a significant lack of immune response, often referred to as 'bubble boy disease'. It details the classification of immunodeficiencies into primary (congenital) and secondary (acquired), as well as various forms of SCID caused by genetic mutations that affect both humoral and cell-mediated immunity. Common clinical features include recurrent infections, failure to thrive, and specific enzyme deficiencies related to disease prognosis.






























































































