Downloaded 281 times

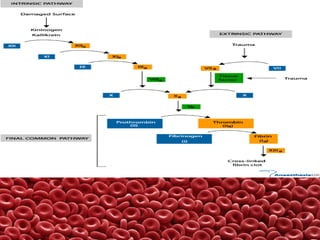

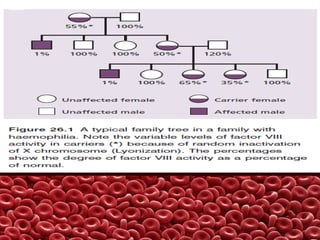
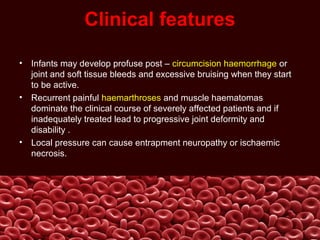
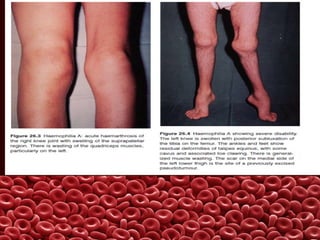
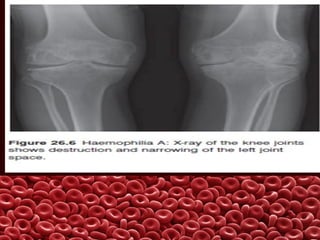
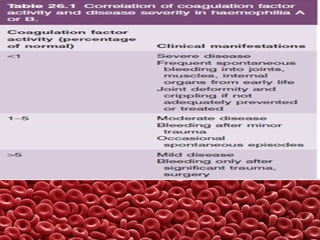

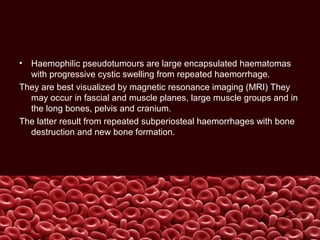

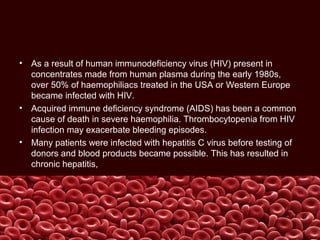

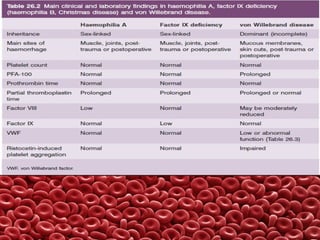





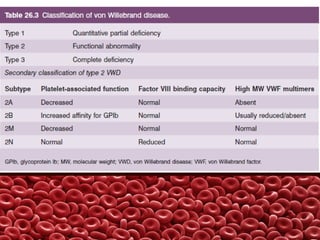
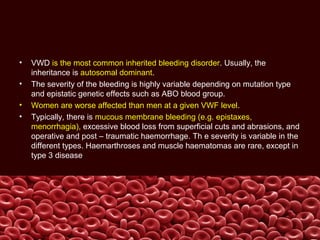

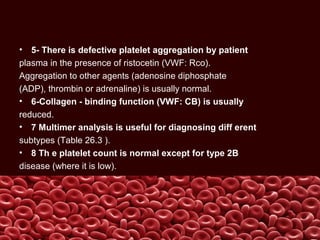
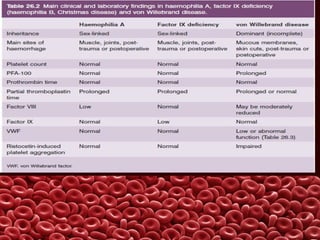












This document summarizes several coagulation disorders including haemophilia A, haemophilia B, von Willebrand disease, other hereditary coagulation factor deficiencies, and disseminated intravascular coagulation. It describes the clinical features, inheritance, laboratory findings, treatment, and management of each condition. Key points include that haemophilia A is the most common clotting factor deficiency, von Willebrand disease is the most common inherited bleeding disorder, and disseminated intravascular coagulation can be triggered by trauma, infection, cancer and other conditions.



































































