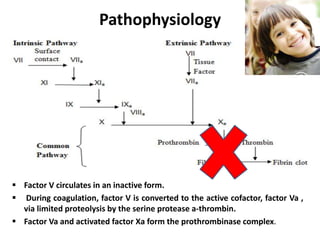
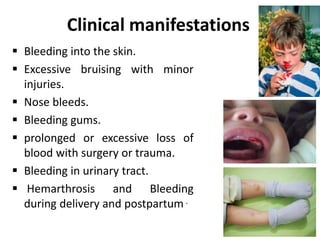
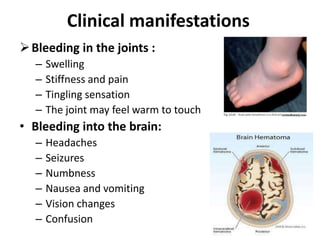
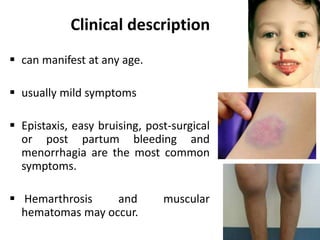
Factor V deficiency, also known as Owren’s disease, is a rare bleeding disorder caused by insufficient or dysfunctional factor V, leading to impaired blood coagulation. It can be inherited (autosomal recessive) or acquired due to immune responses and presents with a range of bleeding symptoms from mild bruising to severe hemorrhaging. Diagnosis is based on laboratory assays showing decreased factor V activity, and treatment involves fresh frozen plasma infusions and monitoring, with no known prevention for the inherited form.











![Laboratory Studies
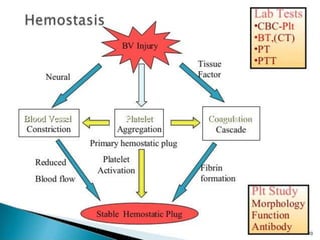
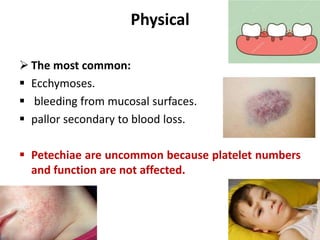
Factor V assay (decreased activity) .
Bleeding time (prolonged in severe case).
aPTT (Prolonged).
PT (Prolonged).
Thrombin time (Normal).
Stypven time (Russell viper venom time [RVVT]):
(Prolonged).
Correction of PT or partial thromboplastin time
(PTT) with the mixing of equal amounts of normal
and patient plasma.](https://image.slidesharecdn.com/factorvdeficiency-200114180910/85/Factor-v-deficiency-20-320.jpg)





































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)








