Downloaded 60 times










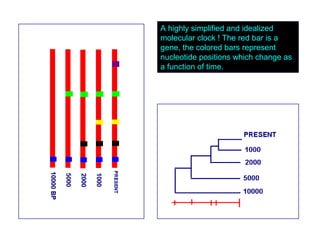


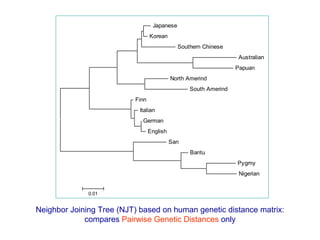

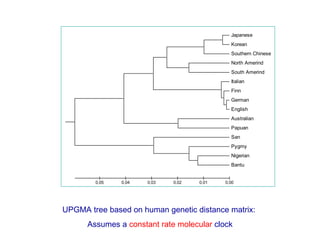









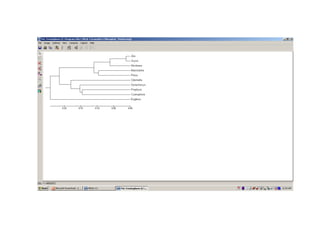






The document introduces phylogenetics and the use of the MEGA 6.0 software for phylogenetic analysis, emphasizing the importance of classification in understanding evolutionary relationships. It discusses traditional and molecular taxonomy, explains species versus gene trees, and highlights various phylogenetic methods and statistical validations such as bootstrapping. The document also outlines the functionalities of MEGA 6.0, its user-friendly features, and the steps to perform phylogenetic analysis.































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)














