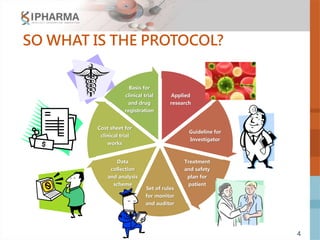
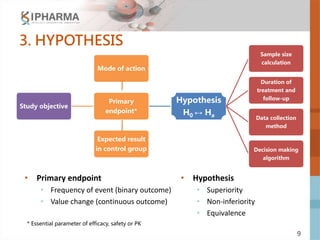
The document outlines the key elements of a successful clinical trial protocol. A protocol is a document that provides guidelines for a clinical trial, including its objectives, design, methodology, and organization. It aims to obtain appropriate data on a drug's clinical properties within the shortest timeline and at minimal cost, while ensuring patient safety and adherence to regulatory standards. The document discusses 10 essential elements for an effective protocol: medical expertise, prior experience, hypothesis, endpoints, safety measures, credibility, rationality, structure, simplicity, and legitimacy. It emphasizes the importance of a protocol in providing clear guidelines to ensure a clinical trial's objectives are achieved efficiently and ethically.



































![Indian gcp guidelines[647]](https://cdn.slidesharecdn.com/ss_thumbnails/indiangcpguidelines647-210325044800-thumbnail.jpg?width=640&height=640&fit=bounds)









































































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)








