The document outlines the ICH GCP E6 guidelines, which serve as a standard for the conduct of clinical trials to ensure data credibility and protect trial subjects' rights. It details the core principles, responsibilities of sponsors and investigators, the informed consent process, and essential documentation required for compliance. Adhering to these guidelines is crucial for maintaining the integrity and ethical standards of clinical research involving human participants.

















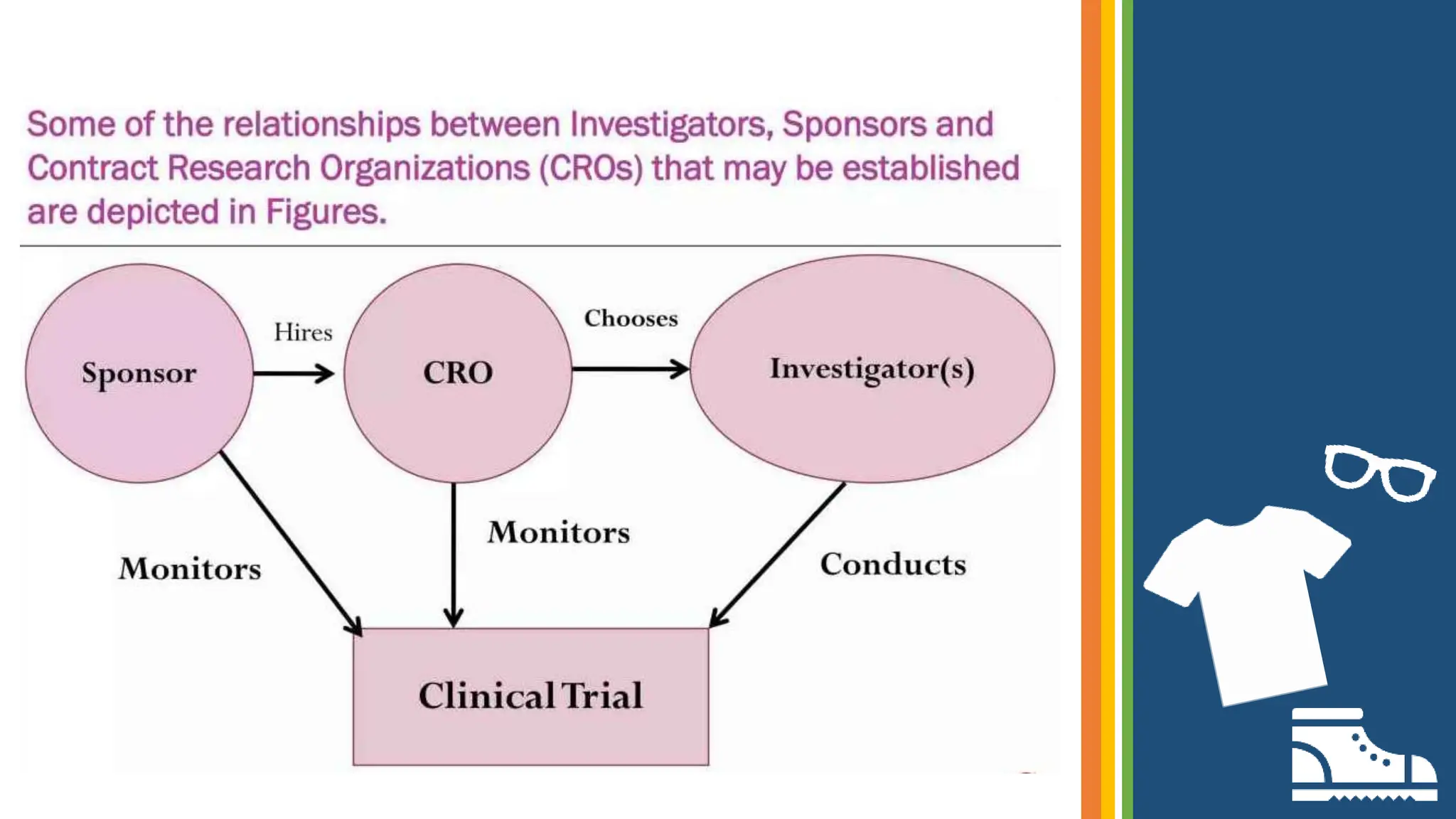















![roles and responsibilities of Investigator[663]](https://cdn.slidesharecdn.com/ss_thumbnails/investigator663-210616055819-thumbnail.jpg?width=640&height=640&fit=bounds)































































