



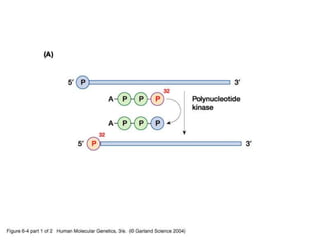
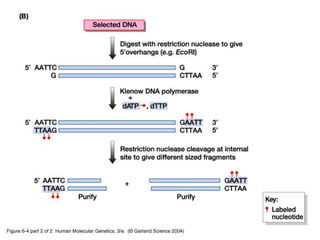

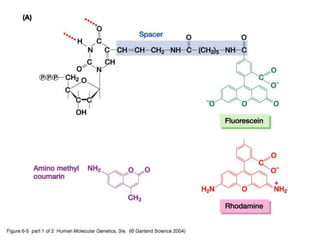
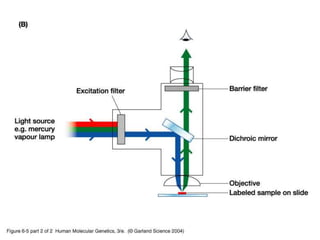




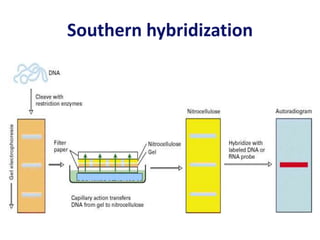
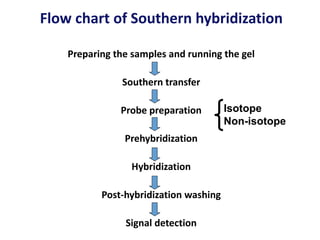







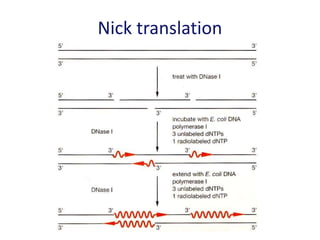

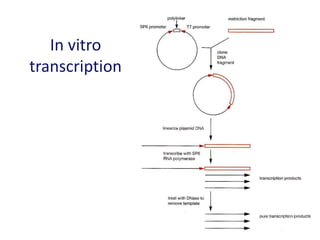


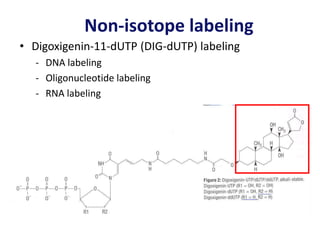
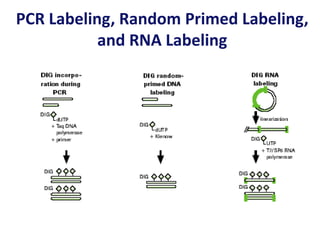



![Critical parameters (II)
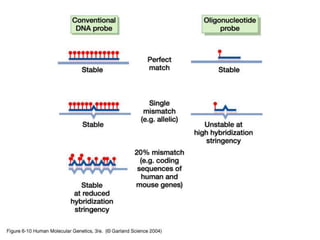
• Homology between the probe and the sequences
being detected
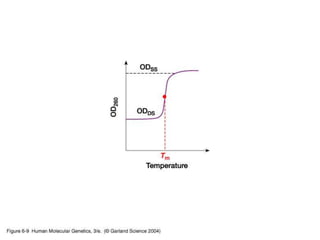
– Tm = 81 +16.6 (log Ci) + 0.4 [% (G+C)] - 0.6 (%
formamide)- 600/n - 1.5 (% mismatch)
– Factors can be changed:
• Hybridization temp.
• Washing temp.
• Salt concentration during washing
High temp., low salt: high stringency
Low temp., high salt: low stringency
– If 50 % formamide is used
• 42 oC for 95 ~ 100 % homology
• 37 oC for 90 ~ 95 % homology
• 32 oC for 85 ~ 90 % homology](https://image.slidesharecdn.com/blotting2017-200730202922/85/Blotting-2017-44-320.jpg)




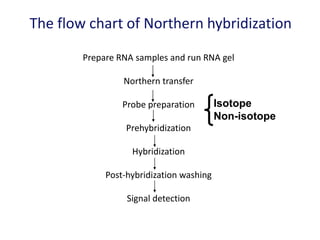



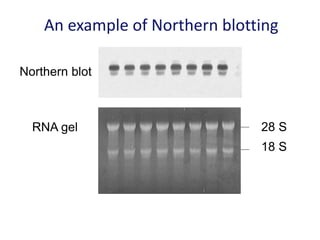


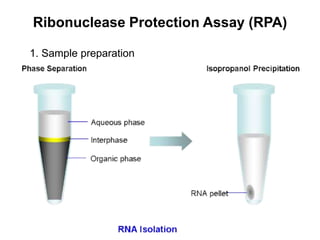
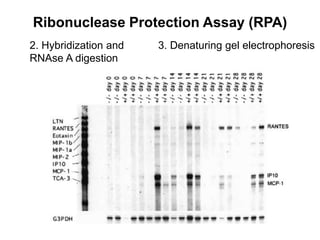

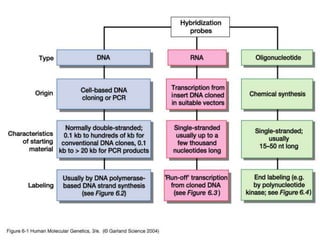
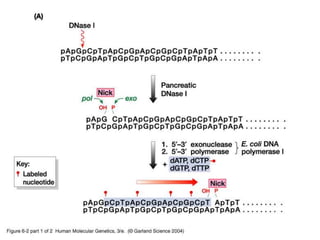
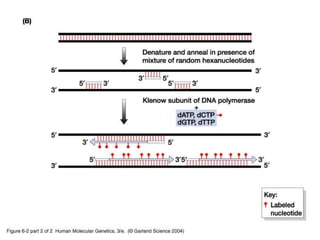
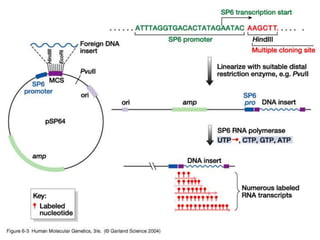
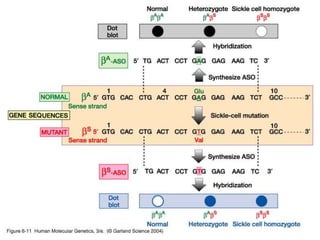
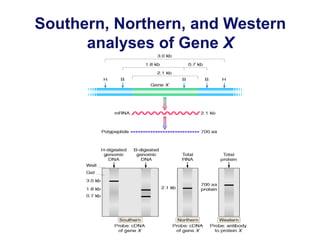
The document discusses various techniques for preparing and labeling nucleic acid probes, including DNA, RNA, and oligonucleotides. Probes can be labeled isotopically through strand synthesis during polymerase reactions or end-labeling, or non-isotopically with modified nucleotides containing fluorophores or affinity molecules. The principles of nucleic acid hybridization, such as factors affecting melting temperature and stringency, are also covered. Common hybridization techniques like Southern blot, Northern blot, and dot-blot are summarized, involving transferring nucleic acids to membranes, hybridizing probes, and detecting signals.

















































































