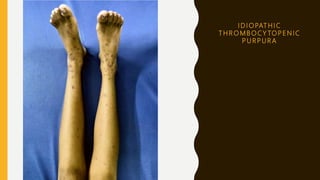
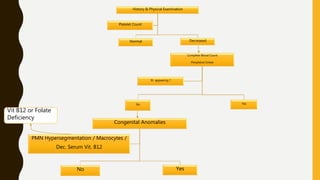
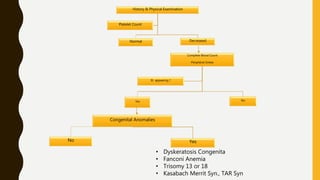
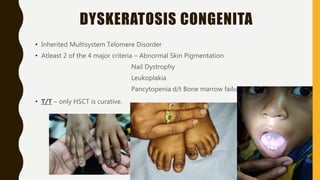
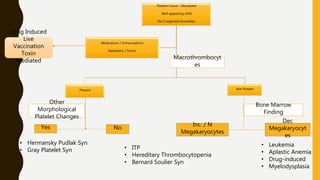
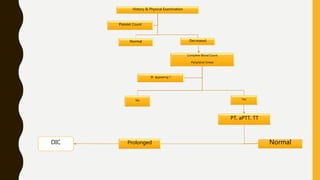
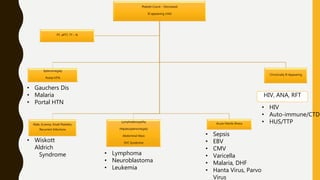
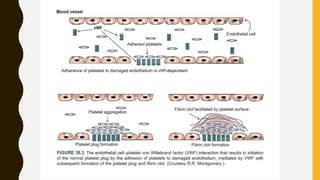
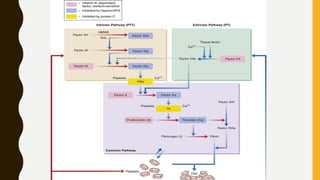
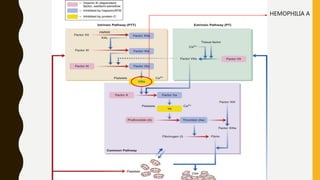
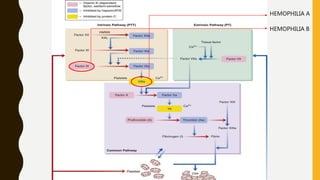
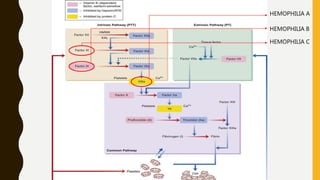
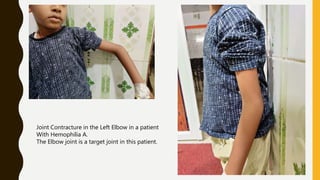
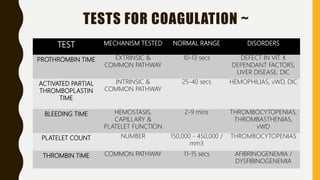
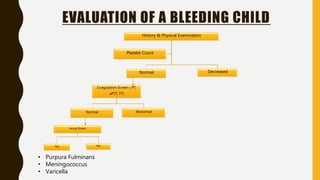
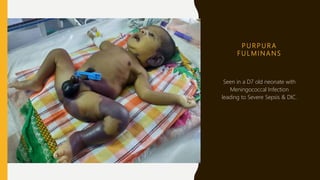
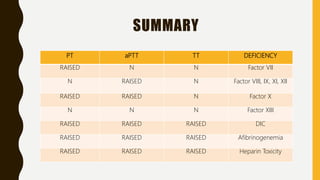
This document discusses the approach to evaluating and treating bleeding in children. It covers the physiology of hemostasis, clinical evaluation, hereditary and acquired bleeding disorders like hemophilia A/B and von Willebrand disease, and platelet disorders. Specific case scenarios are presented to demonstrate how different bleeding patterns and test results can help diagnose conditions like hemophilia or platelet function defects. The role of factor replacement therapies, desmopressin, and other treatments are also summarized.














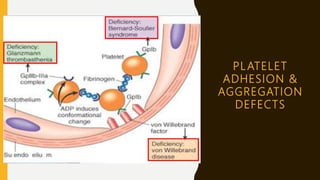


![BERNARD SOULIER SYNDROME
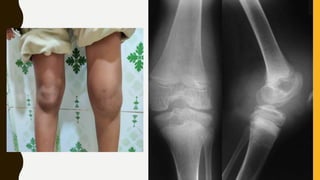
Severe Congenital platelet function disorder
Absence / Severe Def. of receptor for vWF on the platelets [ Gp Ib-IX ]
C/F – Thrombocytopenia
Giant platelets
Greatly prolonged Bleeding Time / PFA-100 Closure Time
Significant mucosal & GI bleeding
Inv – Absent Ristocetin Aggregation. Normal Aggregation with other agonists.
vWF – Normal
Confirmation by – Flow Cytometry of platelet Glycoproteins](https://image.slidesharecdn.com/approachtoableedingchild-191208152202/85/Approach-To-A-Bleeding-Child-38-320.jpg)