Downloaded 293 times







































































![Treatment
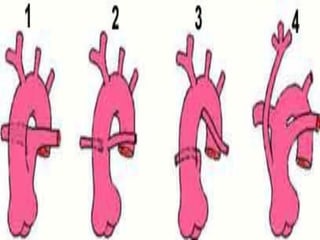
Once symptomatic onset occurs, a common treatment is decompression surgery,[14] in
which a neurosurgeon usually removes the lamina of the first and sometimes the second
or even third cervical vertebrae and part of the occipital bone of the skull to relieve
pressure. The flow of spinal fluid may be accompanied by a shunt. Since this surgery
usually involves the opening of the dura mater and the expansion of the space beneath, a
dural graft is usually applied to cover the expanded posterior fossa.
A small number of neurological surgeons believe that detethering the spinal cord as an
alternate approach relieves the compression of the brain against the skull opening
(foramen magnum), obviating the need for decompression surgery and associated
trauma. However, this approach is significantly less documented in the medical literature,
with reports on only a handful of patients. It should be noted that the alternative spinal
surgery is also not without risk.
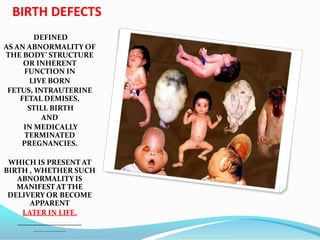
Prognosis
The prognosis differs dependent on the type of malformation (i.e., type I, II, III, or IV). Type
I is generally adult-onset and, while not curable, treatable and non-fatal. Types I and II
sufferers may also develop syringomyelia. Type II is typically diagnosed at birth or
prenatally. Approximately 33% of individuals with Chiari II malformation develop
symptoms of brainstem damage within five years; a 1996 study found a mortality rate of
33% or more among symptomatic patients, with death frequently occurring due to
respiratory failure. 15% of individuals with Chiari II malformation die within two years of
birth. Among children under two who also have myelomeningocele, it is the leading cause
of death. Prognosis among children with Chiari II malformation who do not have spina
bifida is linked to specific symptoms; the condition may be fatal among symptomatic
children when it leads to neurological deterioration, but surgical intervention has shown
promise. Types III and IV are extremely rare and patients generally do not survive past the
age of two or three](https://image.slidesharecdn.com/birthdefect2014-140508063627-phpapp02/85/Birth-defect-2014-75-320.jpg)






































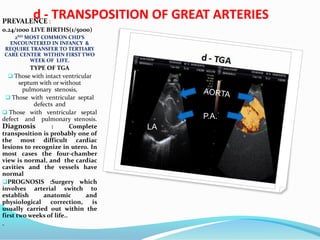





























![CONGENITAL GLUCOMA
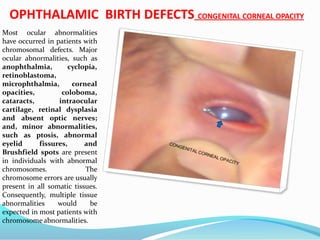
Buphthalmos is defined as a
"large eye" [bu (Greek) = ox or
cow]. It is most often present
in both eyes in children due to
congenital open-angle
glaucoma of the eye, noted by
unusually large corneas and
increased overall size of the
eyeball. An abnormally narrow
angle between the cornea and
iris blocks the outflow of
aqueous humor, which leads to
an increased intraocular
pressure and a characteristic
bulging enlargement of the
eyeball. Patient symptoms may
include excessive tearing and
light sensitivity
("photophobia"). Cupping of
the optic disk, which may be
the first sign to be seen on
dilated examination by an eye
care professional. Congenital
glaucoma untreated usually
leads to blindness.](https://image.slidesharecdn.com/birthdefect2014-140508063627-phpapp02/85/Birth-defect-2014-146-320.jpg)

































































































































































1. The document describes fetal development from pre-embryonic to embryonic stages, covering organogenesis and development of major organs from 4-12 weeks. 2. It then covers types of birth defects such as malformations, disruptions, deformations, and dysplasias which can be caused by genetic or environmental factors such as infections, drugs, or radiation exposure during pregnancy. 3. The major causes of birth defects are described as genetic factors in 40-60% of cases, maternal illnesses and infections in 20-25% of cases, and multifactorial and unknown causes in 12-25% and 10-13% of cases respectively.






































































































