This document outlines the process of constructing phylogenetic trees to delineate relationships among Coronaviridae species using protein sequences. It describes:
1) Choosing nucleocapsid and membrane proteins as molecular markers and collecting sequences from NCBI.
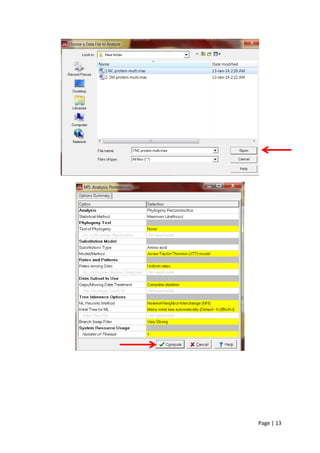
2) Performing multiple sequence alignment on the proteins using MUSCLE in MEGA, which is more accurate than ClustalW.
3) Selecting maximum likelihood as the tree-building method because it uses all sequence information without reducing it to distances and makes fewer assumptions than other methods.