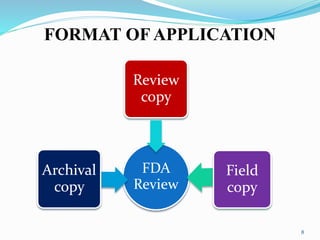
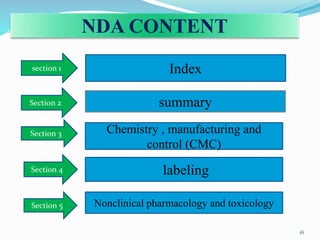
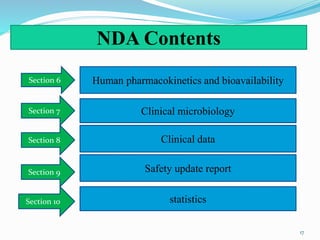
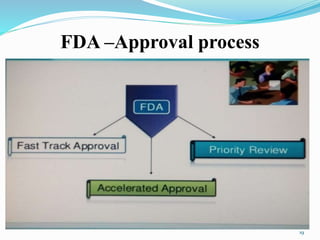
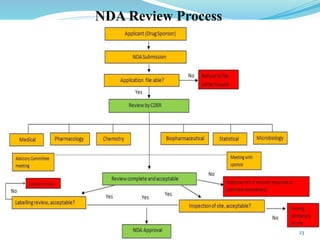
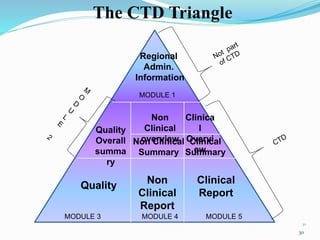
The document discusses the New Drug Application (NDA) process in the United States. It provides an overview of the history and goal of the NDA, the application format and forms, contents of an NDA, and the FDA approval process. The NDA is submitted to the FDA by drug companies to obtain approval to market a new pharmaceutical product. It contains extensive information on the drug's safety, efficacy, manufacturing and quality controls. The FDA review process involves evaluation of the application by medical and regulatory experts to determine if the drug's benefits outweigh its risks.