Download as PDF, PPTX

















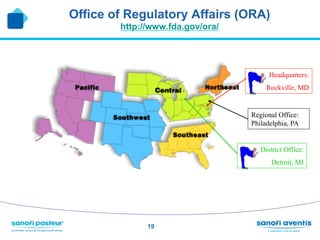

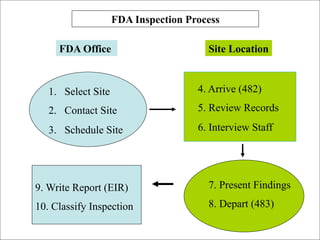




























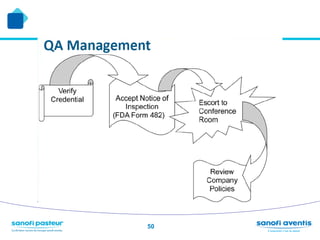










The document discusses preparing for and handling an FDA inspection at a facility. It covers the FDA's authority to inspect facilities and outlines the different types of inspections including routine, concise, follow-up, special, and quality systems reviews. It also discusses the inspection process, providing details on the frequency, duration, and whether inspections are announced or unannounced. Tips are provided on how to prepare for an inspection, what to do during an inspection, and what information is allowed and not allowed to be shared with FDA inspectors.























![Handling of a fda inspection [compatibility mode]](https://cdn.slidesharecdn.com/ss_thumbnails/handlingofafdainspectioncompatibilitymode-150610120404-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)











































