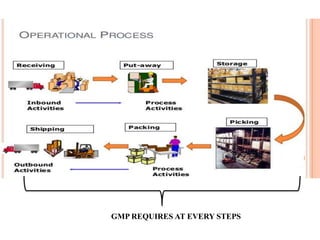
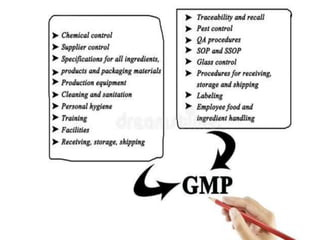
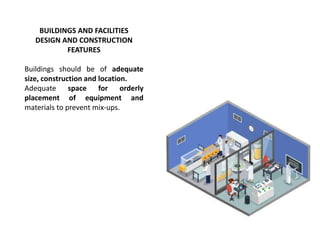
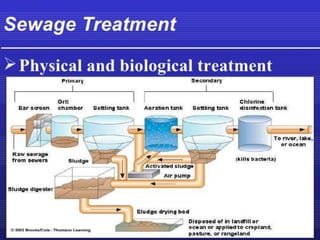
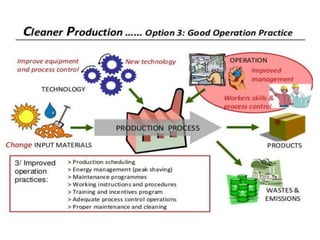
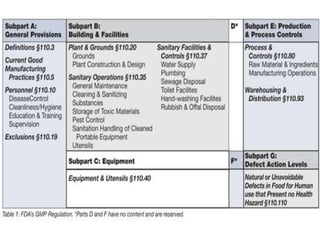
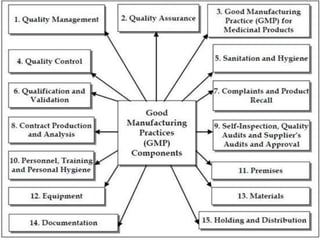
Good manufacturing practices (GMP) regulations ensure consistency and quality in pharmaceutical manufacturing. GMP covers facilities, equipment, personnel, production, packaging and quality control. Key requirements include designated clean areas for operations, qualified personnel, documented procedures, process validation, environmental monitoring, component testing and record keeping. GMP aims to prevent contamination and errors through strict quality standards at all stages of production.