




![ Transition state is not very polar
Not strongly affected by solvent polarity
In most cases, reaction is a concerted [4πs + 2πs]
Cycloaddition](https://image.slidesharecdn.com/13-dipolarcycloaddition-200522152641/75/1-3-dipolar-cycloaddition-Reactions-8-2048.jpg)






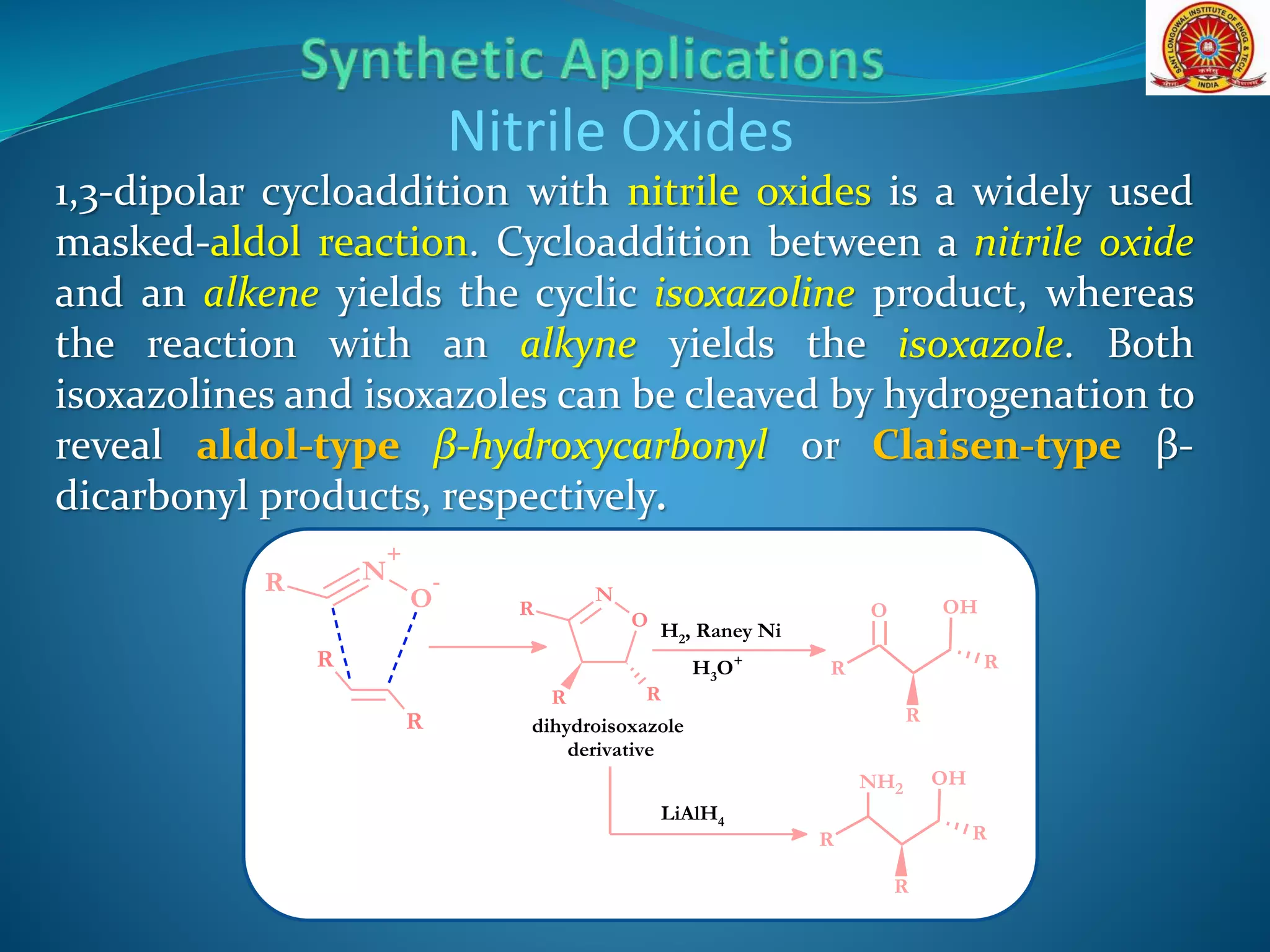
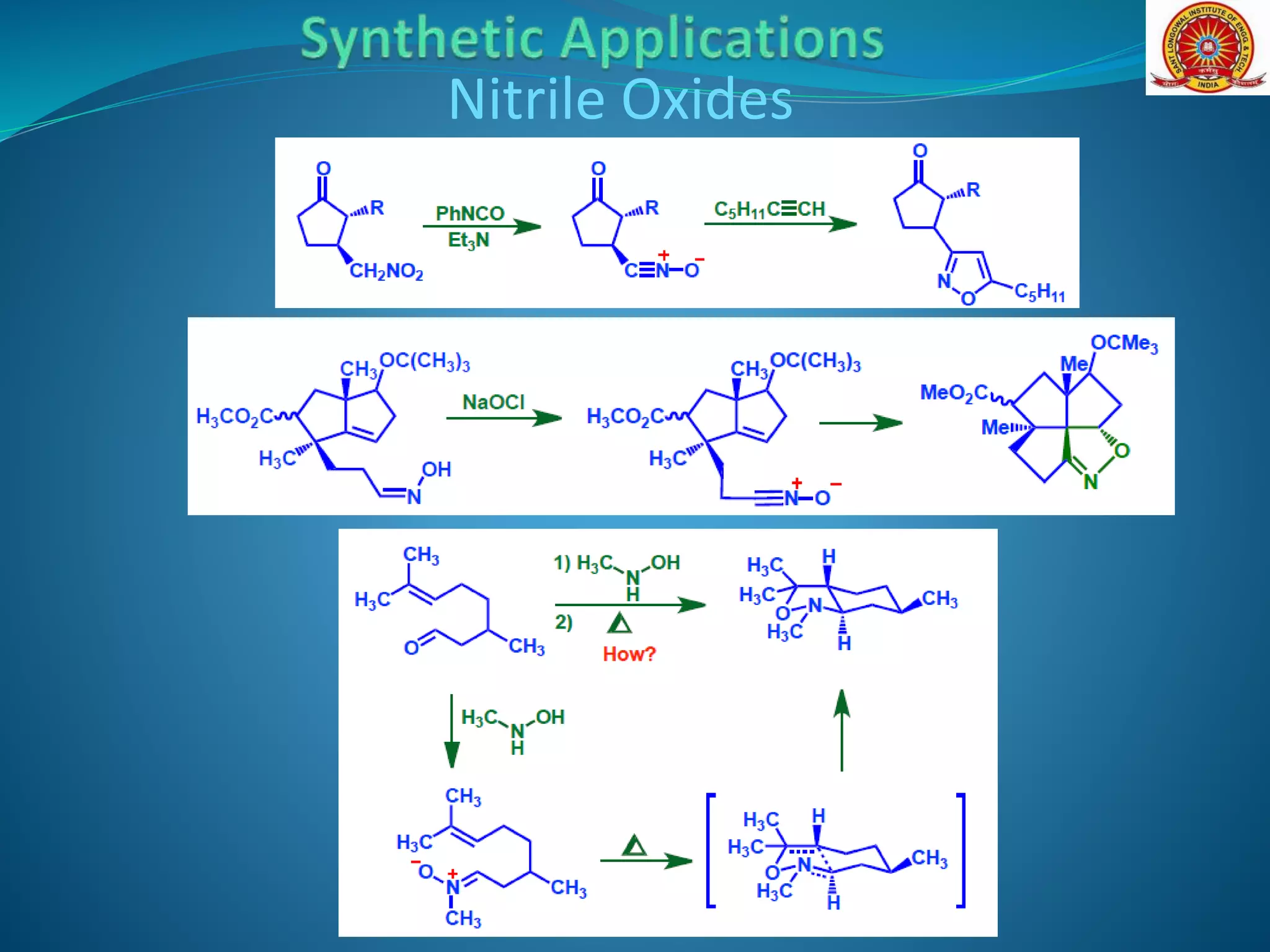
![Nitrile Oxides
Intra-molecular 1,3-dipolar cycloaddition reactions take place
readily and some useful applications of this chemistry in
synthesis have been reported. In the synthesis of the
antitumor agents sarcomycin [D, where R=H], dehydration of
the nitro-alkene (A) gave the isoxazoline (B) via the
intermediate nitrile oxide. Hydrogenolysis with Raney nickel in
aqueous acetic acid then led to the b-hydroxy ketone (C),
which was dehydrated to the ethyl ester of sarcomycin (D), R=
Et
(B)(A) (C) (D)](https://image.slidesharecdn.com/13-dipolarcycloaddition-200522152641/75/1-3-dipolar-cycloaddition-Reactions-20-2048.jpg)



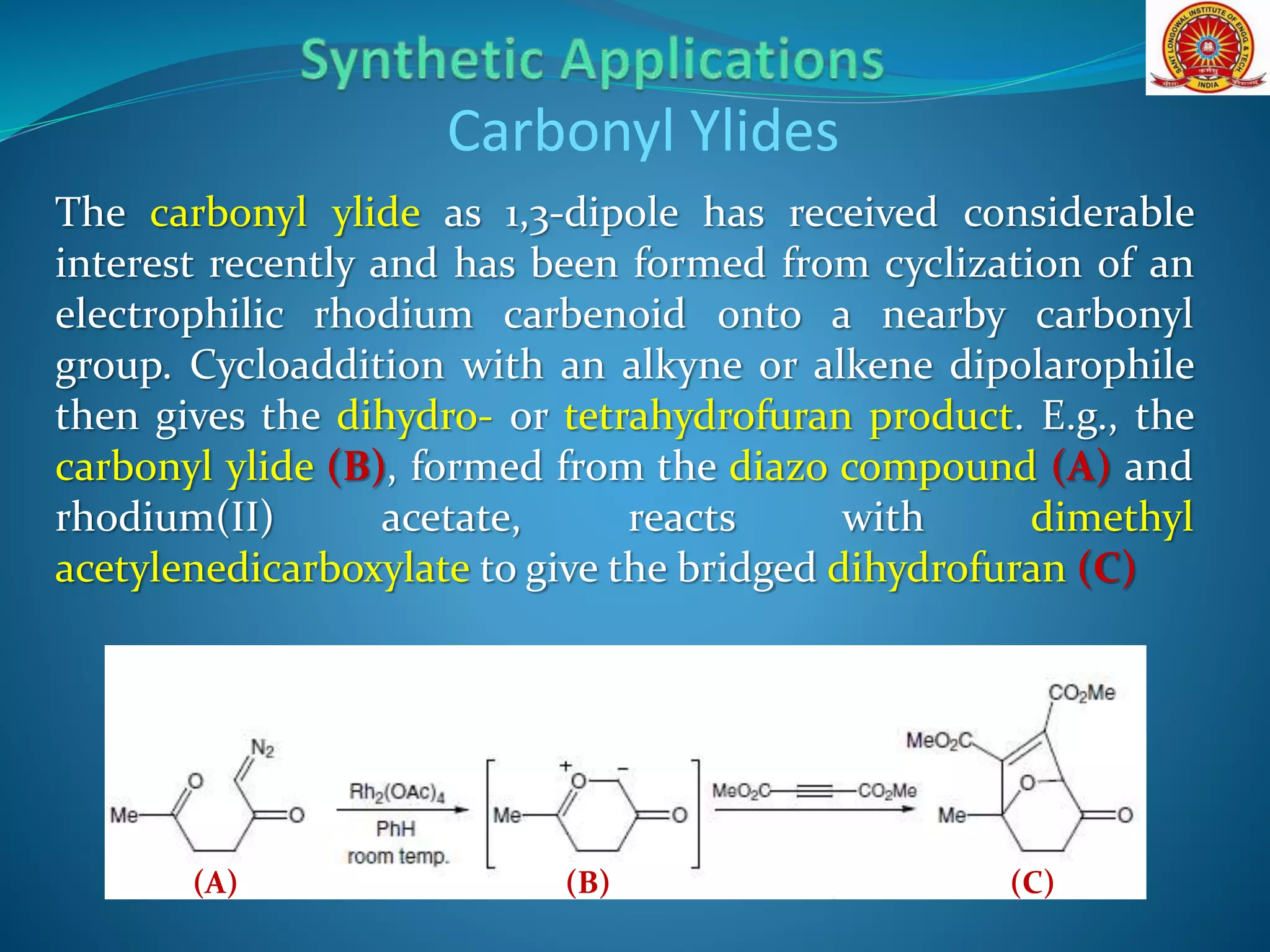




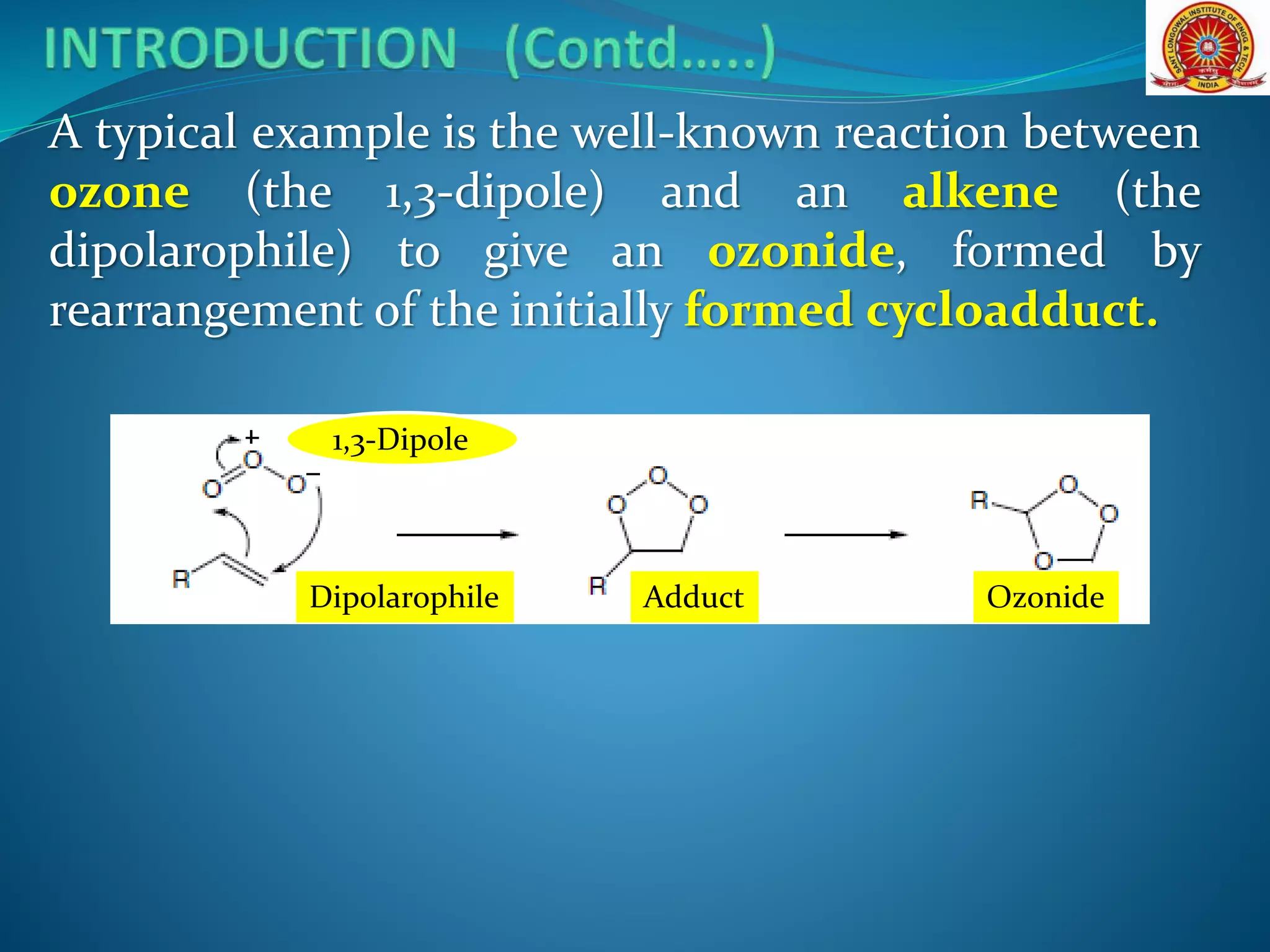
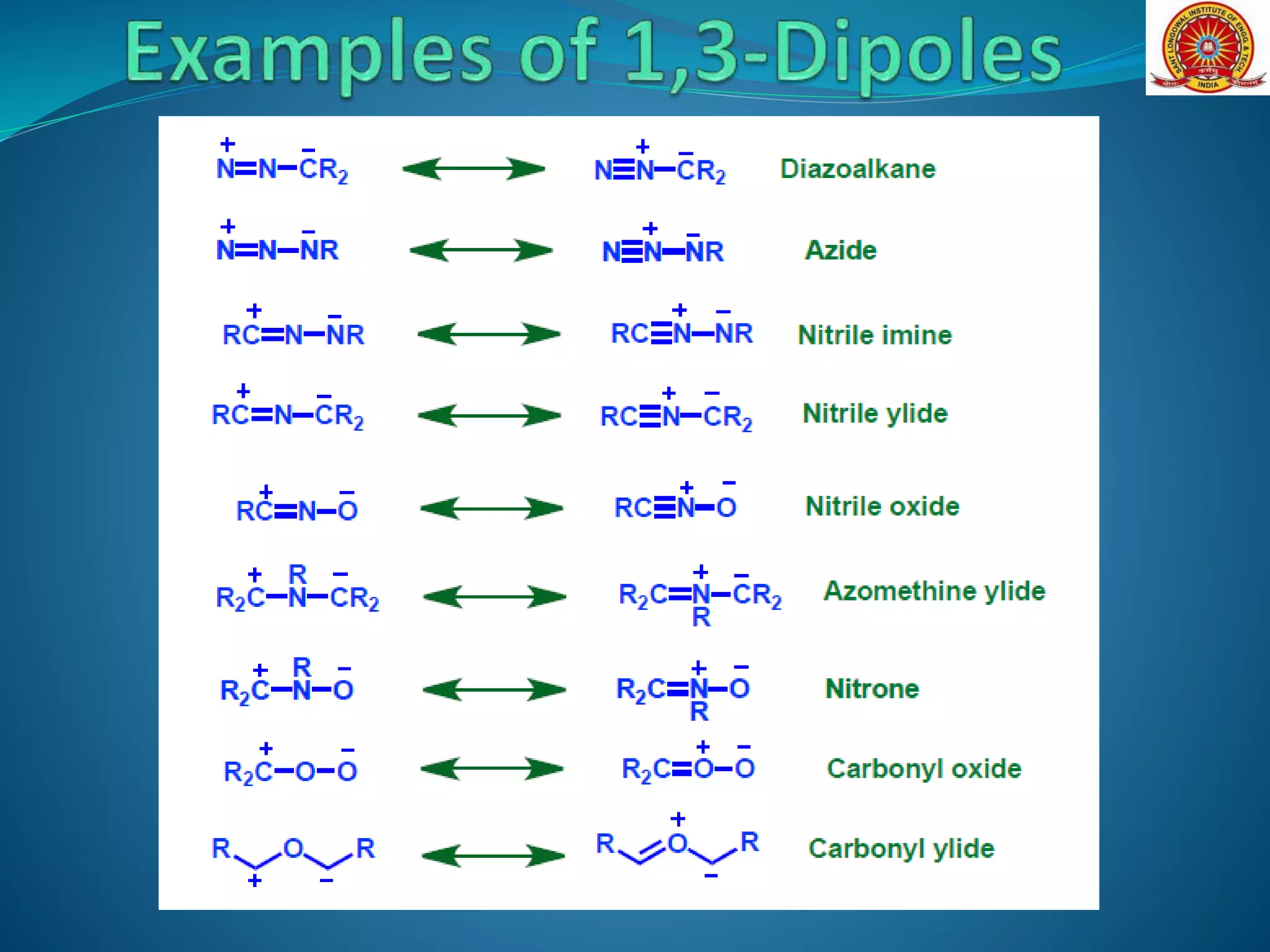
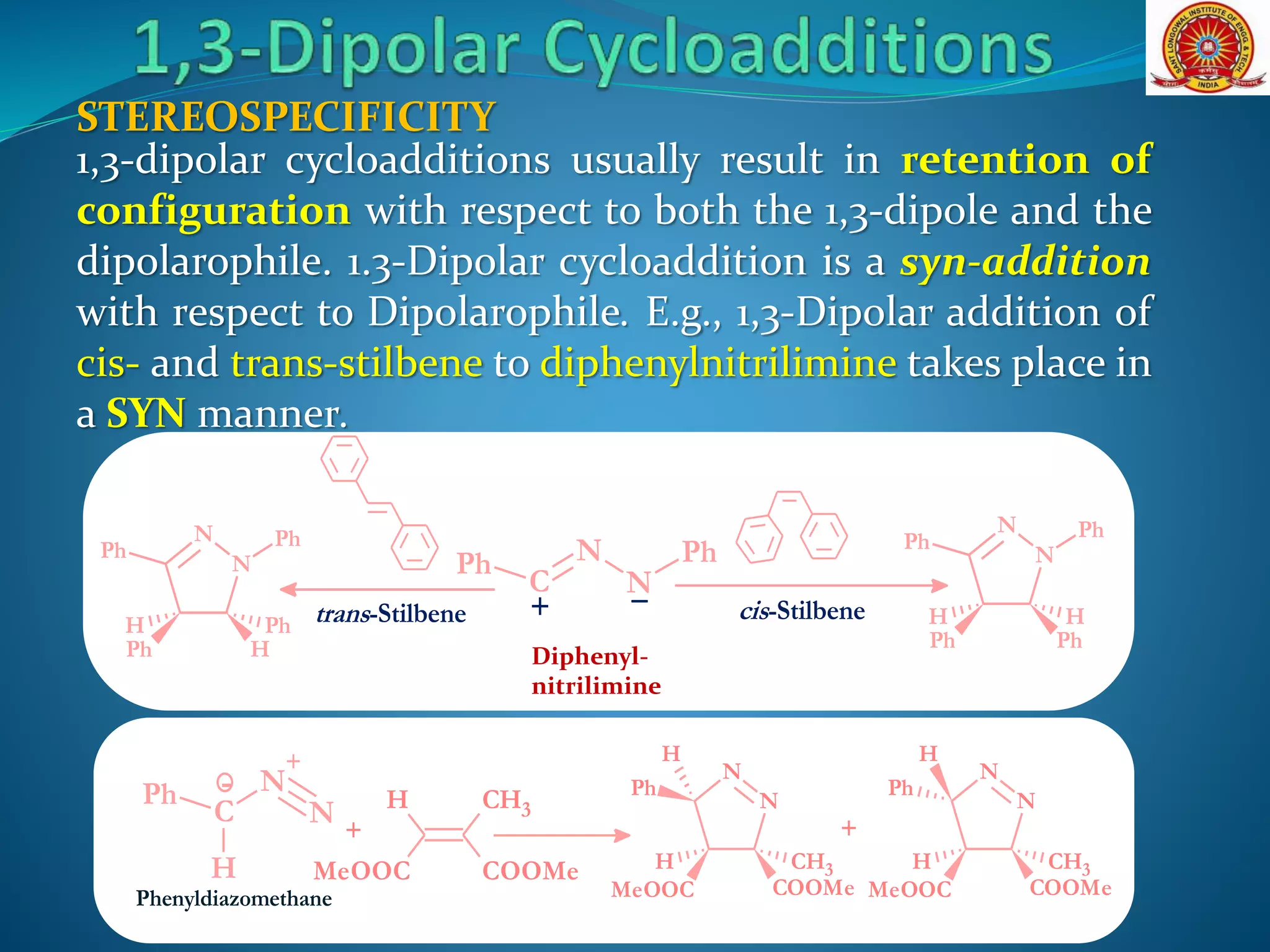
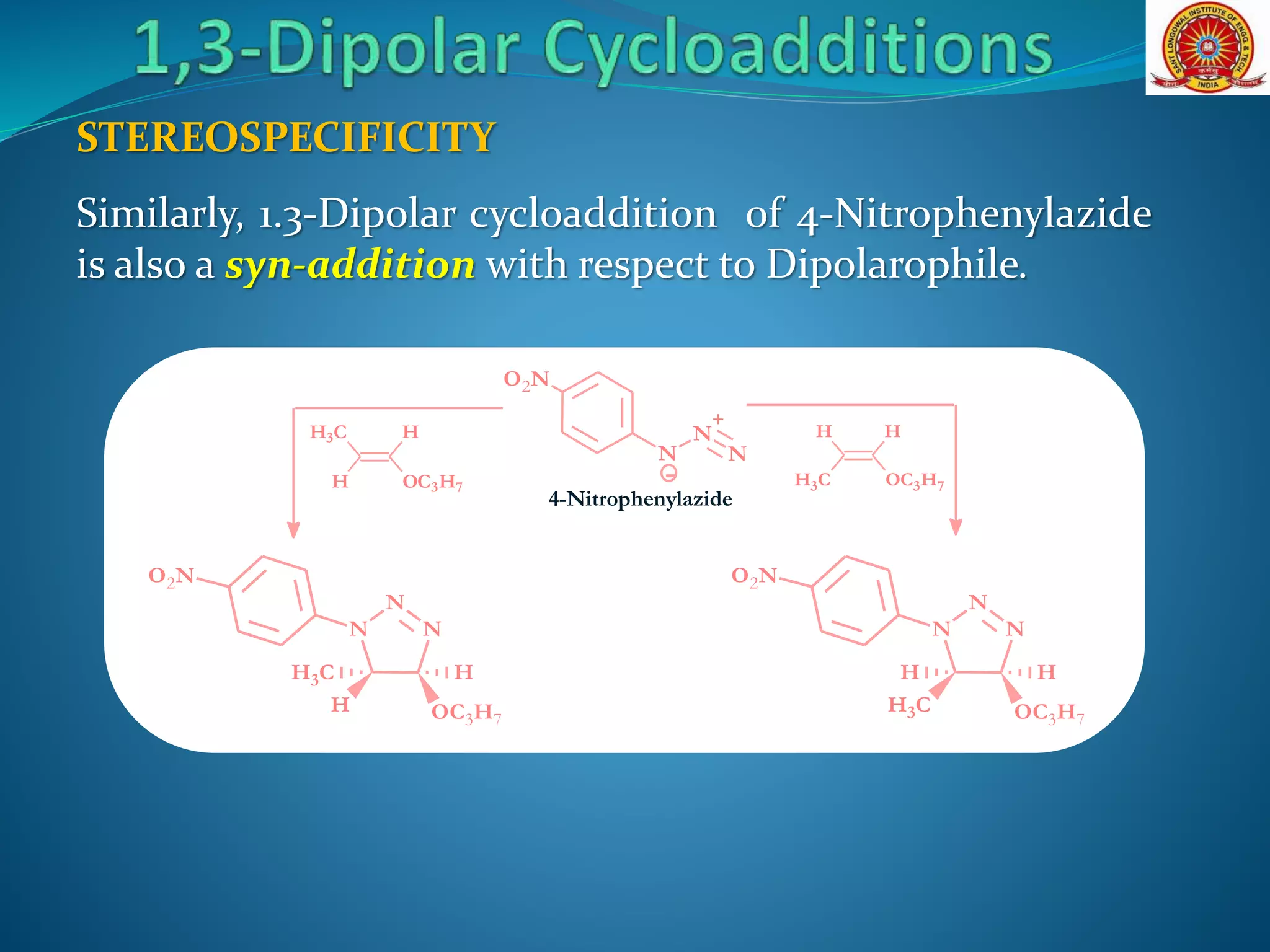
The document discusses 1,3-dipolar cycloaddition reactions, which involve a 1,3-dipole reacting with a dipolarophile to form a 5-membered heterocyclic ring. Key points include: - 1,3-dipoles are classified into three types based on their electronic structure. Common examples are azides, nitrones, and carbonyl ylides. - The reaction typically proceeds by a concerted pericyclic mechanism through a six-electron transition state, though some exceptions involve a stepwise mechanism. - Frontier molecular orbital theory can be used to classify dipoles as HOMO-controlled, LUMO-controlled, or ambiphilic based
















![Cycloaddition reactions [2+2]](https://cdn.slidesharecdn.com/ss_thumbnails/cycloadditionreactions22-200514145354-thumbnail.jpg?width=640&height=640&fit=bounds)




































![Cycloaddition reactions [2+2]](https://cdn.slidesharecdn.com/ss_thumbnails/cycloadditionreactions22-200522152644-thumbnail.jpg?width=640&height=640&fit=bounds)
























