Downloaded 50 times




















![http://biotic.isciii.es/ [email_address] Instituto de Salud Carlos III Medical Bioinformatics Area Thanks ¡¡¡ http://www.inbiomedvision.eu/](https://image.slidesharecdn.com/1130ws20lopez-120119041959-phpapp01/85/INBIOMEDvision-Workshop-at-MIE-2011-Victoria-Lopez-29-320.jpg)

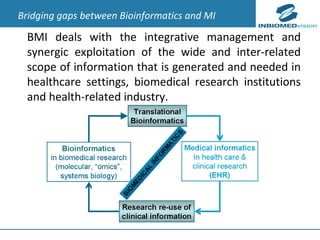
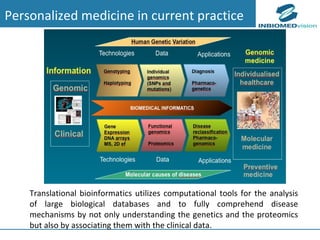
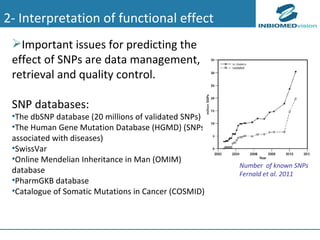
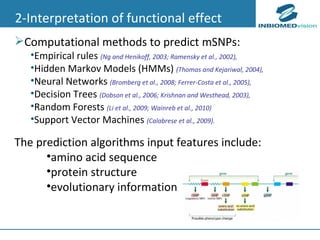
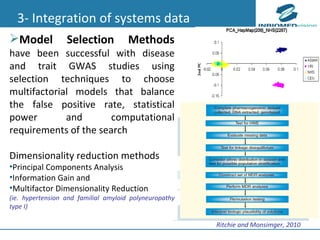
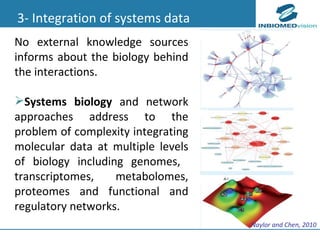
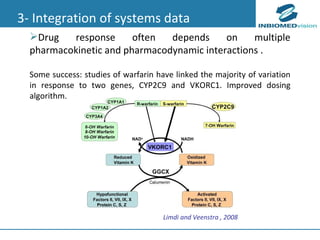
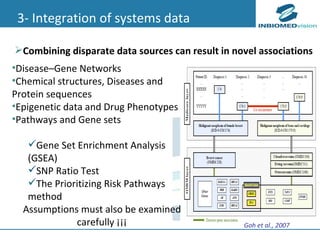
1) Personalized medicine currently faces challenges in processing large-scale genomic data, interpreting the functional effects of genomic variations, integrating systems-level data, and translating discoveries into medical practice. 2) Bioinformatics can help address these challenges through algorithms for mapping and aligning sequencing data, predicting functional effects, prioritizing genes, integrating multi-omics data into networks, and disseminating discoveries through databases to inform medical practice. 3) Fully realizing personalized medicine will require overcoming limitations of current approaches, validating computational predictions, and updating medical practice and education to routinely incorporate genomic information.

























































































![5G Explained! A High Level Overview [Introduction]](https://cdn.slidesharecdn.com/ss_thumbnails/5gexplainedahighleveloverview-260119165306-cc137a3e-thumbnail.jpg?width=640&height=640&fit=bounds)



