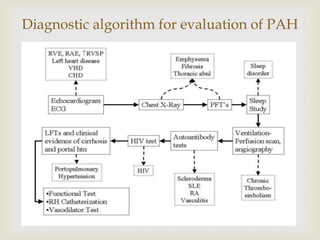
This document discusses pulmonary arterial hypertension (PAH), including its definition, classification, pathogenesis, clinical manifestations, natural history, diagnosis, and management. PAH is defined as a sustained elevation of pulmonary arterial pressure over 25 mm Hg at rest or 30 mm Hg with exercise, caused by various genetic and environmental factors. It is classified into 5 groups by the WHO based on underlying mechanisms. Common symptoms include dyspnea and fatigue. Treatment includes anticoagulants, vasodilators like prostacyclins, endothelin receptor blockers, PDE-5 inhibitors, and in severe cases, lung transplantation.




![
Imbalance of vascular effectors
Associated environmental factors:hypoxia,
anorexigens[fenfluramine,dexfenfluramine],CNS
stimulants[methamphetamine,cocaine]
Other associated conditions: scleroderma, (HIV),
human herpesvirus (HHV), portal hypertension,
thrombocytosis, hemoglobinopathies, and hereditary
hemorrhagic telangiectasia.
Associated genetic abnormalities:
Pathogenesis](https://image.slidesharecdn.com/primarypulmonaryhypertension-130912101111-phpapp01/85/Primary-pulmonary-hypertension-5-320.jpg)