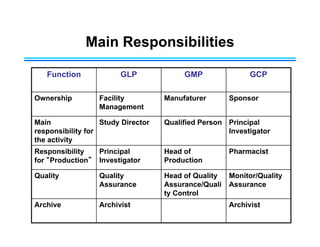
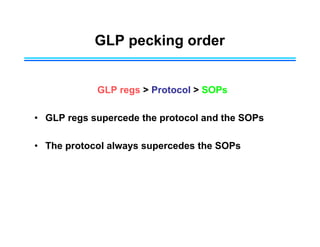
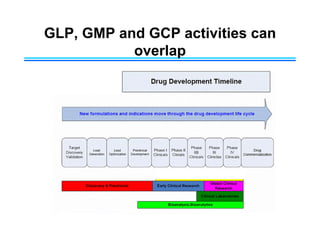
This document provides an overview of key differences between Good Laboratory Practices (GLP), Good Manufacturing Practices (GMP), and Good Clinical Practices (GCP). GLPs regulate non-clinical safety studies and ensure reliability and integrity of test data. GMPs govern pharmaceutical manufacturing and quality control. GCPs provide standards for clinical trials involving human subjects to protect rights and welfare. While the principles have overlapping goals of quality and compliance, they apply to distinct phases of research and product development.



























































































![Good_Laboratory_Practice-(GLP)_VJ[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/goodlaboratorypractice-vjedited1-251101061707-c8f8e4cf-thumbnail.jpg?width=640&height=640&fit=bounds)













